GO富集分析柱状图
target_gene_id <- unique(read.delim("miRNA-gene interactions.txt")$EntrezID)
# BiocInstaller::biocLite("clusterProfiler")
# BiocInstaller::biocLite("org.Hs.eg.db")
display_number = c(, , )
## GO enrichment with clusterProfiler
library(clusterProfiler)
ego_MF <- enrichGO(OrgDb="org.Hs.eg.db",
gene = target_gene_id,
pvalueCutoff = 0.05,
ont = "MF",
readable=TRUE)
ego_result_MF <- as.data.frame(ego_MF)[:display_number[], ]
# ego_result_MF <- ego_result_MF[order(ego_result_MF$Count),]
ego_CC <- enrichGO(OrgDb="org.Hs.eg.db",
gene = target_gene_id,
pvalueCutoff = 0.05,
ont = "CC",
readable=TRUE)
ego_result_CC <- as.data.frame(ego_CC)[:display_number[], ]
# ego_result_CC <- ego_result_CC[order(ego_result_CC$Count),]
ego_BP <- enrichGO(OrgDb="org.Hs.eg.db",
gene = target_gene_id,
pvalueCutoff = 0.05,
ont = "BP",
readable=TRUE)
ego_result_BP <- na.omit(as.data.frame(ego_BP)[:display_number[], ])
# ego_result_BP <- ego_result_BP[order(ego_result_BP$Count),]
go_enrich_df <- data.frame(ID=c(ego_result_BP$ID, ego_result_CC$ID, ego_result_MF$ID),
Description=c(ego_result_BP$Description, ego_result_CC$Description, ego_result_MF$Description),
GeneNumber=c(ego_result_BP$Count, ego_result_CC$Count, ego_result_MF$Count),
type=factor(c(rep("biological process", display_number[]), rep("cellular component", display_number[]),
rep("molecular function", display_number[])), levels=c("molecular function", "cellular component", "biological process")))
## numbers as data on x axis
go_enrich_df$number <- factor(rev(:nrow(go_enrich_df)))
## shorten the names of GO terms
shorten_names <- function(x, n_word=, n_char=){
if (length(strsplit(x, " ")[[]]) > n_word || (nchar(x) > ))
{
if (nchar(x) > ) x <- substr(x, , )
x <- paste(paste(strsplit(x, " ")[[]][:min(length(strsplit(x," ")[[]]), n_word)],
collapse=" "), "...", sep="")
return(x)
}
else
{
return(x)
}
}
labels=(sapply(
levels(go_enrich_df$Description)[as.numeric(go_enrich_df$Description)],
shorten_names))
names(labels) = rev(:nrow(go_enrich_df))
## colors for bar // green, blue, orange
CPCOLS <- c("#8DA1CB", "#FD8D62", "#66C3A5")
library(ggplot2)
p <- ggplot(data=go_enrich_df, aes(x=number, y=GeneNumber, fill=type)) +
geom_bar(stat="identity", width=0.8) + coord_flip() +
scale_fill_manual(values = CPCOLS) + theme_bw() +
scale_x_discrete(labels=labels) +
xlab("GO term") +
theme(axis.text=element_text(face = "bold", color="gray50")) +
labs(title = "The Most Enriched GO Terms")
p
pdf("go_enrichment_of_miRNA_targets.pdf")
p
dev.off()
svg("go_enrichment_of_miRNA_targets.svg")
p
dev.off()
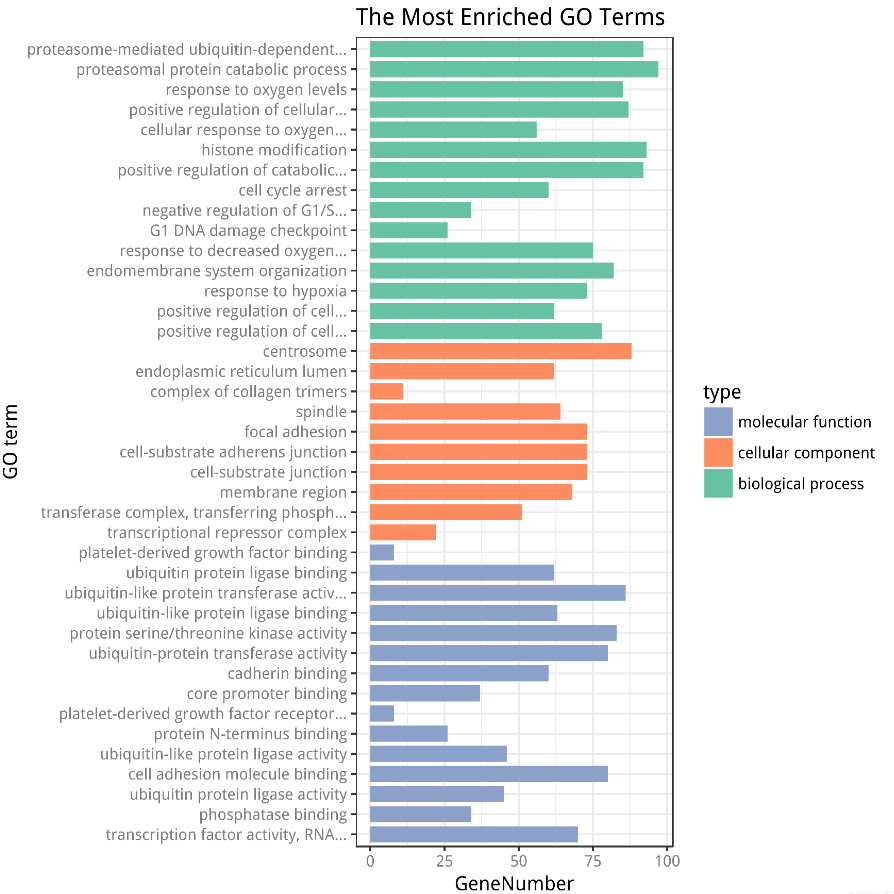
GO富集分析柱状图的更多相关文章
- 基因探针富集分析(GSEA)& GO & pathway
http://blog.sina.com.cn/s/blog_4c1f21000100utyx.html GO是Gene Ontology的简称,是生物学家为了衡量基因的功能而而发起的一个项目,从分子 ...
- GO富集分析示例【华为云技术分享】
版权声明:本文为博主原创文章,遵循CC 4.0 BY-SA版权协议,转载请附上原文出处链接和本声明. 本文链接:https://blog.csdn.net/devcloud/article/detai ...
- DAVID 进行 GO/KEGG 功能富集分析
何为功能富集分析? 功能富集分析是将基因或者蛋白列表分成多个部分,即将一堆基因进行分类,而这里的分类标准往往是按照基因的功能来限定的.换句话说,就是把一个基因列表中,具有相似功能的基因放到一起,并和生 ...
- 利用GSEA对基因表达数据做富集分析
image Gene Set Enrichment Analysis (GSEA) is a computational method that determines whether an a p ...
- R: 修改镜像、bioconductor安装及go基因富集分析
1.安装bioconductor及go分析涉及的相关包 source("http://bioconductor.org/biocLite.R") options(BioC_mirr ...
- GO富集分析
GO的主要用途之一是对基因组进行富集分析.例如,给定一组在特定条件下上调的基因,富集分析将使用该基因组的注释发现哪些GO术语被过度表示(或未充分表示). 富集分析工具 用户可以直接从GOC网站的 ...
- OS Tools-GO富集分析工具的使用与解读详细教程
我们的云平台上的GO富集分析工具,需要输入的文件表格和参数很简单,但很多同学都不明白其中的原理与结果解读,这个帖子就跟大家详细解释~ 一.GO富集介绍: Gene Ontology(简称G ...
- webgestalt 通路富集分析
http://www.webgestalt.org/ 通路富集分析 参考 http://www.sci666.com.cn/9596.html
- GSEA 基因集富集分析
http://software.broadinstitute.org/gsea/index.jsp GSEA(Gene Set Enrichment Analysis)是一种生物信息学的计算方法,用于 ...
随机推荐
- CSMA/CD 3
一.二进制指数类型退避算法 (truncated binary exponential type) 发生碰撞的站在停止发送数据后,要推迟(退避)一个随机时间才能再发送数据. 目的:重传时再次发生碰撞的 ...
- 组合数计算-java
排列组合是计算应用经常使用的算法,通常使用递归的方式计算,但是由于n!的过于大,暴力计算很不明智.一般使用以下两种方式计算. 一,递归的思想:假设m中取n个数计算排列组合数,表示为comb(m,n). ...
- 编程题A+B Format的总结(第二次作业<一>)
Github链接地址:https://github.com/Startup-try/object-oriented 这个题目现在想想没有那么难,其实还挺简单的,但是中午花了好长的时间还不懂得怎么做,感 ...
- Mininet自定义网络拓扑
在Mininet上的网络拓扑有两种方式 第一种 用mininet自带的miniedit可视化工具,在mininet/mininet/examples/的目录下的一个miniedit.py,运行这个文件 ...
- DIY简单功能的torrentkitty种子爬虫
过完年回公司比较无聊,一不小心看到微博里美尤莉娅的图片,惊为天人,有图为证!!! 百度之原来这货以前叫小泉彩,貌似动了几个小手术换了个马甲重新出道了.你拍AV你家里知道么?.于是乎下了几个种子看了下, ...
- CSS加载性能优化
将首屏页面要用到的CSS文件,放在页面头部加载,其他模块的CSS可以使用异步加载:loadCSS 和 Preload. 关于preload,推进2篇文章给大家看下: 1.通过rel="pre ...
- css计数器 及 鼠标经过从中间扩散一个矩形(正方形长方形均可)
<!DOCTYPE html> <html> <head> <title>css计数器--兼容IE8</title> <meta ch ...
- 【[APIO2007]动物园】
我好\(sb\)啊,把\(>>\)打成\(<<\)结果就写了两节课 那个一个人只能看到五个动物显然很鬼畜 那我们就可以压这一维了 \(dp[i][s]\)表示从第\(i\)个位 ...
- urllib下使用Xpath表达式示例
urllib下使用Xpath表达式示例 使用xpath表达式需要先将需要匹配的数据转换成tree格式,这就需要先装lxml模块.安装方法可以使用pip安装. 示例代码: import urllib.r ...
- 用firefox的插件下载网页中的视频
对于网页中的一些视频,直接下载不了,可以用专用下载软件下载,也可以用firefox的NetVideohunter Video Downloader插件下载网页中的视频,方便快捷. 工具/原料 fi ...