FastQC 测序质量
文章转载于 Original 2017-07-06 Jolvii 生信百科
介绍一下如何理解 FastQC 各模块的结果
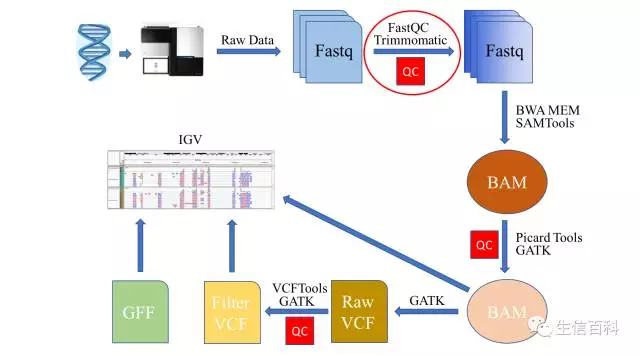
FastQC 的使用
FastQC的安装介绍请看这里。FastQC 支持 fastq、gzip 压缩的 fastq、SAM、BAM 等格式,在不指定文件类型的情况下,FastQC 会根据文件的名字来推测文件的类型: 以 .sam 或者 .bam 结尾的文件会被当作 SAM/BAM 文件来打开,并统计 mapped 和 unmapped reads 在内的所有 reads;其它的文件类型则被当作 fastq 格式打开。 其使用语法为:
fastqc [-o output dir] [--(no)extract] [-f fastq|bam|sam] [-c contaminant file] [-t threads] seqfile1 .. seqfileN
-o 用来指定输出文件的目录,需要注意的是,FastQC 不会自动创建新目录,故指定的目录必须存在;
FastQC 输出结果为 .zip 文件,默认参数为 --extract (自动解压缩),执行时加上 --noextract 则不解压缩;
-f 用来指定输入文件格式,如果不指定则自动检测;
-c 用来指定一个文件,这个文件里面存放可能存在的污染序列,FastQC 会在这个文件里面搜索 reads 中的 overrepresented sequences;
-t 用来指定同时处理的文件个数;
seqfile1 等是需要处理的文件名称;
详细信息请见 fastqc -h 或者 fastqc --help;
我用来分析的命令为 fastqc --noextract -t 2 sample_R1.fq.gz sample_R2.fq.gz -o ./00.FastQC
FastQC 结果解读
上一期我简单地提了一下 FastQC 结果的基本内容,在思考之后,决定把结果中每部分内容给大家仔细解读一下。如图 2 所示:FastQC 的结果包含 12 个方面,其中绿色的结果表示“通过”;黄色的结果表示“警告”;红色的结果表示“不合格”。我们应关注结果中未通过的部分,仔细思考为什么我们的数据会得到这样的结果,可能存在哪些问题?下面我们分别看一下各部分结果的内容,以及 FastQC 判断各部分结果通过、警告和不合格的阈值是什么

1. Basic Statistics
Basic Statistics 的结果给出原始数据的基本信息,包括被分析文件的文件名、文件类型 (actual base calls/colorspace data)、质量值编码方式、序列总数、标记为低质量的序列数、序列长度 和 GC 含量,如图 3 所示:
图 3 Basic Statistics
Basic Statistics 的状态始终都是“通过”,不会出现“警告”或者“不合格”;
这部分结果提供了碱基质量值编码方式,可以记录下来,在后续的分析中会用到。
2. Per Base Sequence Quality
Per Base Sequence Quality 显示 fastq 文件内每一个位置上 (x 轴) 所有碱基的质量值范围 (y 轴),如图 4 所示:
图 4 Per Base Sequence Quality
图中每一位置都有一个 BoxWhisker 图: 黄色箱子表示 25 - 75 % 的范围,即 IQR (inter-quartile range),下面和上面的触须分别表示 10 % 和 90 % 的点。蓝线表示均值,红线表示中位数;
碱基的质量值越高越好,背景颜色将图分成三部分:碱基质量很好 (绿色)、碱基质量一般(黄色) 以及碱基质量差 (红色)。
如果任何一个位置的下四分位数小于10或者中位数小于25,会显示“警告”;如果任何一个位置的下四分位数小于5或者中位数小于20,会显示“不合格”。
3. Per Tile Sequence Quality
只有在分析 Illumina 测序结果且保留了序列 ID 信息 (@HWI-D00523:75:C4PY7ANXX:2:1101:1316:2178,见上一讲 fastq 格式介绍) 时才会有这部分结果出现。为了更好的理解这部分的内容,我先简单的介绍一下 flow cell 的构成 (图 5): 图中所示的 flow cell 有八个 lane (lane1 - lnae8),每个 lane 里面有 3 列 (column1 - column3),每一列内有100 个 tiles,每个 tile 里面又有 20000 - 30000 个 clusters (不同型号 flow cell 内的 column 数、tile 数及 cluster 数会有一定的差异)。
图 5 flow cell 的构成
Per Tile Sequence Quality 的结果展示每个 tile 在每个碱基位置上偏离这个位置所有 tiles 平均质量值的情况,如图 6 所示:
图 6 Per Tile Sequence Quality
图中横轴代表碱基位置,纵轴代表 tile 编号;
图中的颜色是从冷色调到暖色调的渐变,冷色调表示这个 tile 在这个位置上的质量值高于所有 tile 在这个位置上的平均质量值,暖色调表示这个 tile 的在这个位置上的质量值比其它 tiles 要差;
一个很好的结果,整张图都应该是蓝色;
如果任何 tile 的平均质量值与这个位置上所有 tiles 的平均质量值相差 2 以上会显示“警告”,如果任何 tile 的平均质量值与这个位置上所有 tiles 的平均质量值相差 5 以上会显示“不合格”。
4. Per Sequence Quality Scores
Per Sequence Quality Scores 显示每条序列平均碱基质量的分布,如图 7 所示:

图中横轴为测序质量值,纵轴为 reads 数量;
由于成像的原因,得到的测序结果中通常会出现某些 reads 的质量值偏低,这样低质量的 reads 会在图中出现另外一个峰。本图显示的是一个较好的测序结果;
如果最高峰的质量值小于 27 (错误率 0.2 %) 则会显示“警告”,如果最高峰的质量值小于 20 (错误率 1 %) 则会显示“不合格”。
5. Per Base Sequence Content
Per Base Sequence Content 显示每个位置上的碱基组成比例,如图 8 所示:
图 8 Per Base Sequence Content
图中横轴为碱基位置,纵轴为碱基组成比例;
一个完全随机的文库内每个位置上 4 种碱基的比例应该大致相同,因此图中的四条线应该相互平行且接近;
在 reads 开头出现碱基组成偏离往往是我们的建库操作造成的,比如建 GBS 文库时在 reads 开头加了 barcode;barcode 的碱基组成不是均一的,酶切位点的碱基组成是固定不变的,这样会造成明显的碱基组成偏离;
在 reads 结尾出现的碱基组成偏离,往往是测序接头的污染造成的;
如果任何一个位置上的 A 和 T 之间或者 G 和 C 之间的比例相差 10 % 以上则报“警告”,任何一个位置上的 A 和 T 之间或者 G 和 C 之间的比例相差 20 % 以上则报“不合格”。
6. Sequence GC Content
Per Sequence GC Content 显示每条序列平均 GC 含量的分布,如图 9 所示:图 9 Per Sequence GC Content
在一个正常的随机文库中,GC 含量的分布应接近正态分布,且中心的峰值和所测基因组的 GC 含量一致。由于软件并不知道所测物种真实的 GC 含量,图中的理论分布是基于所测数据计算得来的;
如果出现不正常的尖峰分布 (如本图),则说明文库可能有污染 (如果是接头的污染,那么在 overrepresented sequences 那部分结果还会得到提示),或者存在其它形式的偏选;
如果偏离理论分布的 reads 数超过总 reads 数的 15 % 则报“警告”,如果偏离理论分布的 reads 数超过总 reads 数的 30 % 则报“不合格”。
7. Per Base N Content
Per Base N Content 显示每个位置上的 N 的比例,如图 10 所示:
图 10 Per Base N Content
在测序仪工作过程中,如果不能正常完成某个碱基的 calling,将会以 N 来表示这个位置的碱基,而不是 A、T、C、G;
有时在序列中会出现较低比例的 Ns,尤其是靠近序列末端的位置,这说明系统不能正常的 call 这部分碱基;
出现一定比例的 Ns 最常见的原因是普遍出现的质量丢失 (a general loss of quality),这种情况可结合其它部分的结果来综合判断;
另一种常见的现象是文库整体上的测序质量较高,但 reads 开头出现较高比例的 N,这可能是由于文库的碱基组成偏离的比较严重,测序仪不能给出正确的 call,这种情况可以结合 per-base sequence content 的结果来判断;
如果任何一个位置 N 的比例大于 5 % 则报“警告”,大于 20 % 则报“失败”。
8 Sequence Length Distribution
Sequence Length Distribution 的结果显示 reads 长度的分布情况,如图 11 所示:
图 11 Sequence Length Distribution
测序仪出来的原始 reads 通常是均一长度的,但经过质控软件等处理过的数据则不然;
当 reads 长度不一致时报“警告”,当有长度为 0 的 reads 时则报“不合格”。
FastQC 测序质量的更多相关文章
- Fastqc 碱基质量分布图
横坐标代表每个每个碱基的位置,反映了读长信息,比如测序的读长为150bp,横坐标就是1到150: 纵坐标代表碱基质量值, 图中的箱线图代表在每个位置上所有碱基的质量值分布, 中间的红线代表的是中位数 ...
- 测序数据质控-FastQC
通常我们下机得到的数据是raw reads,但是公司通常会质控一份给我们,所以到很多人手上就是clean data了.我们再次使用fastqc来进行测序数据质量查看以及结果分析. fastqc的操作: ...
- 【转录组入门】3:了解fastq测序数据
操作:需要用安装好的sratoolkit把sra文件转换为fastq格式的测序文件,并且用fastqc软件测试测序文件的质量 作业:理解测序reads,GC含量,质量值,接头,index,fastqc ...
- RNAseq测序reads定位
RNAseq测序reads定位 发表评论 3,210 A+ 所属分类:Transcriptomics 收 藏 获得RNA-seq的原始数据后,首先需要将所有测序读段通过序列映射(mapping) ...
- 测序中Q20 Q30 Q40
你能给别人讲清楚这个概念吗? 二代测序中,每测一个碱基会给出一个相应的质量值,这个质量值是衡量测序准确度的.碱基的质量值13,错误率为5%,20的错误率为1%,30的错误率为0.1%.行业中Q20与Q ...
- RNA-seq简单处理流程
RNA_seq pipline RNA_seq pipline PeRl 2018年3月7日 首先说明一下我做RNA-seq处理流程的文件树格式: RNA-seq/ data/ GRCh38.gtf ...
- 一次rna-seq的过程-知乎live转
数据分析流程 来自知乎孟浩巍的“快速入门生物信息学的”Live,超棒的~ 1.数据质控 首先是质控部分,使用fastqc进行对结果分析. 对于Illumia二代测序的结果质控包括两个方面,去掉测序质量 ...
- 生物结构变异分析软件meerkat 0.189使用笔记(二)
一. 运行meerkat 前面已经依序安装了meerkat 的环境和meerkat,运行了预处理一步,在相对应的bam文件目录下生成了大批文件,因此,当要用meerkat处理某个bam文件时,应先将该 ...
- 生物结构变异分析软件meerkat 0.189使用笔记(一)
一.准备工作 meerkat 0.189版本和以前的版本相比,支持bwa mem 输出的bam文件,还支持全外显子数据count SV. meerkat原理:参见http://compbio. ...
随机推荐
- 用angularjs的$http提交的数据,在php服务器端却无法通过$_REQUEST/$_POST获取到
- Zabbix server 3.2安装部署
zabbix server 前提环境: CentOS 6 Lnmp php需要的包(bcmath,mbstring,sockets,gd,libxml,xmlwriter,xmlreader,ctyp ...
- 【paper】KDD15 - Interpreting Advertiser Intent in Sponsored Search
Interpreting Advertiser Intent in Sponsored Search 主要内容是搜索广告的相关性预估模型,使用learning to rank的方法.亮点在于使用了 ...
- C# 根据日期计算星期几
region 根据年月日计算星期几(Label2.Text=CaculateWeekDay(,,);) /// <summary> /// 根据年月日计算星期几(Label2.Text=C ...
- NOI 模拟赛
T1 Article 给 $m$ 个好串,定义一个字符串分割方案是好的当且仅当它分割出来的子串中"是好串的子串"的串长占原串串长超过 85%,定义一个好的分割方案的权值为这种分割方 ...
- 20179223《Linux内核原理与分析》第三周学习笔记
测试3的实验: 1. 用gcc -g编译vi输入的代码 2. 在main函数中设置一个行断点 3. 在main函数增加一个空循环,循环次数为自己学号后4位,设置一个约为学号一半的条件断点 4. 提交调 ...
- ThinkPHP5 使用create 获取表单所有字段
TP5没有 TP3的那个create创建表单字段,如果字段太多,写起来是非常麻烦 只需要在 框架里面 think/db/Query.php 里面加上函数 public function create( ...
- eclipse adt调试出错,不能产出apk问题
The connection to adb is down, and a severe error has occured http://blog.csdn.net/h7870181/article/ ...
- [LeetCode系列]链表环探测问题II
给定一个链表头, 探测其是否有环, 如果没有返回NULL, 如果有返回环开始的位置. 环开始的位置定义为被两个指针指向的位置. 算法描述: 1. 快慢指针遍历, 如果到头说明无环返回NULL, 如果相 ...
- IMP-00013: 只有 DBA 才能导入由其他 DBA 导出的文件
IMP-00013: only a DBA can import a file exported by another DBA 处理方法:在给目标环境的用户赋予dba权限,或者细粒度一些,赋予imp_ ...