R数据分析:孟德尔随机化实操
好多同学询问孟德尔随机化的问题,我再来尝试着梳理一遍,希望对大家有所帮助,首先看下图1分钟,盯着看将下图印在脑海中:
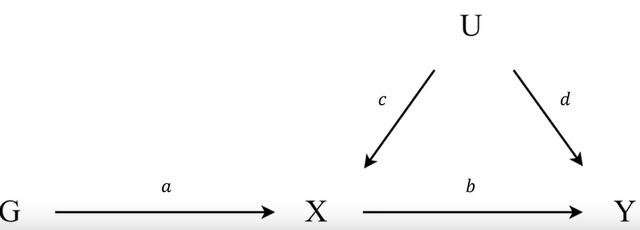
上图是工具变量(不知道工具变量请翻之前的文章)的模式图,明确一个点:我们做孟德尔的时候感兴趣的是x和y的关系,也就是小b,但是我们直接去跑x对y的回归肯定是不对的,因为有很多的U,因此我们借助工具变量G(关于工具变量我们之前的文章有详细的解释,请自行查阅),去估计我们感兴趣的小b。
现在有天然良好的工具变量G,也就是我们的基因变量,此时有上面的图,再次重申:我们感兴趣的,最终希望得到准确估计的值是小b,按照上图我们应该有GY的关系是ab,GX的关系是a,于是乎b可以写成ab/a,就是我们感兴趣的b可以换一种思路得到,如下:

上面的式子要跑通的话,我们需要知道G-Y的关系和G-X的关系。
但是我们GY也就是基因和结局的关系已经有人给我们研究好了,我们可以直接去GWAS里面找研究好的summarydata拿来用就行。
但是我们的的GX也就是基因和暴露的关系也已经有人给我们研究好了,我们可以直接去GWAS里面找研究好的summarydata拿来用就行。
也就是说,通过孟德尔随机化,我们完全可以毫不费力地估计出我们需要的小b,也就是暴露和结局的关系----就是今天要再次给大家介绍的孟德尔随机化研究。
思路就是这么清晰。就是这么清晰。搞不明白的同学再多读几遍。
术语解析
为了帮助大家理解思想,在孟德尔随机化的实操中有几个术语得提点一波:
连锁不平衡(linkage disequilibrium):刚刚讲我们可以有很多的基因结局/暴露的关系的,就是GWAS里面好些基因可以用,这个时候我们不希望基因之间有相关(会造成double counting,使得结果偏倚):
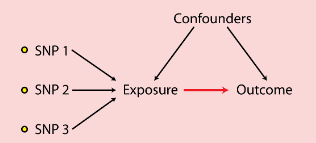
我们实际做的时候,模式是像上图,snp之间你说不相干就不相干?当两个位点的不同等位基因的关联频率高于或低于独立随机关联的条件下的期望频率,这种情况是客观存在的,此时时这些工具变量之间相关性就叫连锁不平衡,其大小可以用LD r方来表示,这个指标也是我们在操作时需要设定的指标之一。
水平基因多效性(Horizontal Pleiotropy):理解这个概念先看下图:
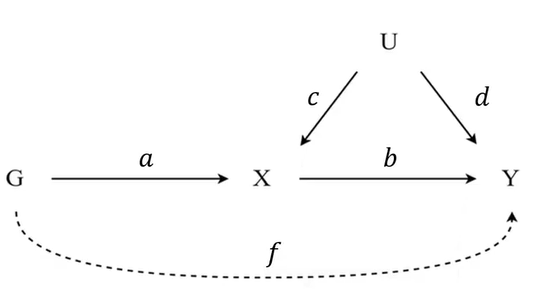
意思是我的理想的情况是通过ab/a的操作估计出b,但是看上图,是不是免不了会出现f这条路径,如果出现了f,我们的基因和结局之间的关系就是f+ab,此时,我用原来的方法估计的就不是b了,而是b+f/a了,就不对了(始终记住我们关心的是b)。
但是如果我的基因变量很多,从而有很多的f,如果所有f的期望均值为0,那么最后我们汇总一下得到的结果也基本上就是b了,无伤大雅。但是就怕所有的f都是一边偏向的(都大于0或都小于0),此时就有问题了,叫做定向多效性directional pleiotropy,这也是为什么我们最后要做漏斗图的原因。
就是通过漏斗图一看都是所有的工具变量都是呈漏斗分布的,就说明没有偏向,这个时候我们认为定向多效性都被冲掉了,不影响。
好,解释了上面的一些术语之后,我们实操一波。
实操
最基本的例子:BMI on CHD的例子,我想看一下BMI作为暴露,CHD作为结局的mr,代码就4条:
bmi_exp_dat <- extract_instruments(outcomes = 'ieu-a-2')
chd_out_dat <- extract_outcome_data(snps = bmi_exp_dat$SNP, outcomes = 'ieu-a-7')
dat <- harmonise_data(bmi_exp_dat, chd_out_dat)
res <- mr(dat)结果如下,下图中有不同方法出来的我们关心的小b:
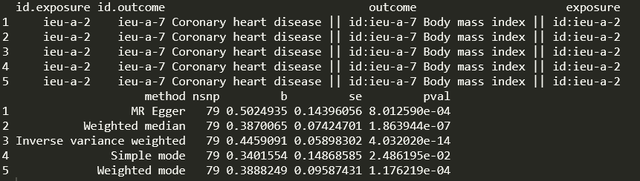
这个就算做完了,就这么简单快速。
接下来就是敏感性分析,首先是各个工具变量的异质性检验:
mr_heterogeneity(dat)运行代码后可以得到Cochran’s Q统计量
然后是水平基因多效性检验,代码如下:
mr_pleiotropy_test(dat)运行代码可以得到egger_intercept
然后是单个SNP结果检验,代码如下:
res_single <- mr_singlesnp(dat)运行后可以得到每个SNP的小b
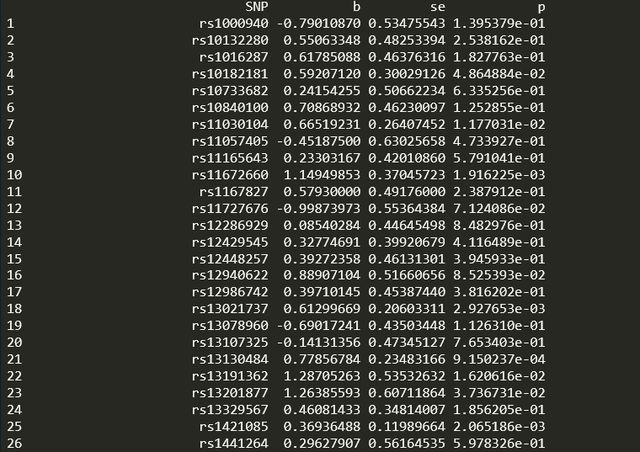
然后是留一检验,代码如下:
mr_leaveoneout(dat)接下来,论文中还会有几个图,首先是点图,代码如下:
mr_scatter_plot(res, dat)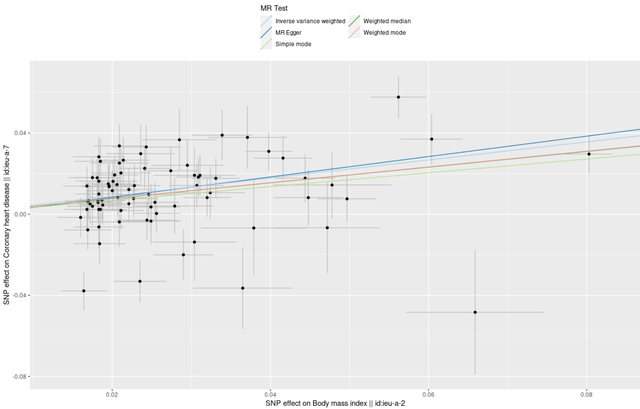
点图是将同一个SNP对暴露的效果放在横轴,对结局的效果放在纵轴,此时图中的斜率就是我们的估计的小b。
然后是单个SNP效应组合的森林图用mr_forest_plot函数可以得到,mr_leaveoneout_plot可以得到留一分析的森林图,mr_funnel_plot可以帮我们得到漏斗图。
到这就出了所有需要报告的东西,做完了。
但是上面的流程有很多的前提,比如你得知道暴露和结局的GWASid才能进行下去,GWAS又有很多,比如你直接用上面的代码的话其实是MR Base GWAS catalog里面的GWAS,当然你还可以选别的,或者用自己找来的最新的GWAS都是可以的。

第一步首先是在相应的GWAS中找到暴露的summary data:
那么有那些GWAS可以供我们使用呢?我们可以直接把GWAS的目录调出来瞅瞅,代码如下:
data(gwas_catalog)运行后大约可以得到15万个全基因组关联研究的数据,截图如下:
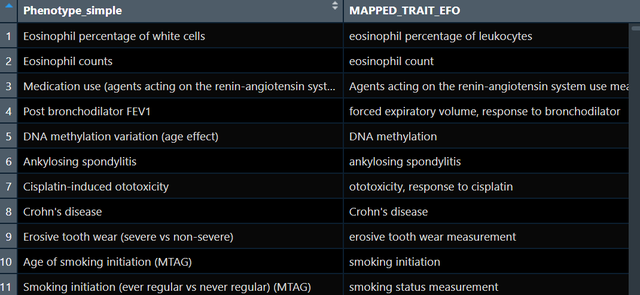
那么对我们而言,我们现在需要找到我们关心的暴露对应的GWAS,比如我现在要找与“blood”表型相关的GWAS,我可以写出如下代码:
exposure_gwas <- subset(gwas_catalog, grepl("Blood", Phenotype_simple))上面的代码相当于只用Phenotype_simple这一列做筛选,当然你也可以结合其它的列比如人群,比如作者,比如地区等等,都是可以的。
选好暴露相关的GWAS之后要做的就是进一步确定基因工具变量和暴露的强度,在论文中一般是这么描述:First, relevance assumption was met considering that all SNPs have reached genome-wide significance (p < 5 × 10−8)
具体的操作如下:
exposure_gwas<-exposure_gwas[exposure_gwas$pval<5*10^-8,]通过上面的步骤保证我们的基因工具变量一定是和暴露强相关。
然后就是将准备好的暴露的GWAS数据形成可以用来做MR分析的数据格式,需要用到format_data()函数:
exposure_data<-format_data(exposure_gwas)此时的exposure_data大概长这样:
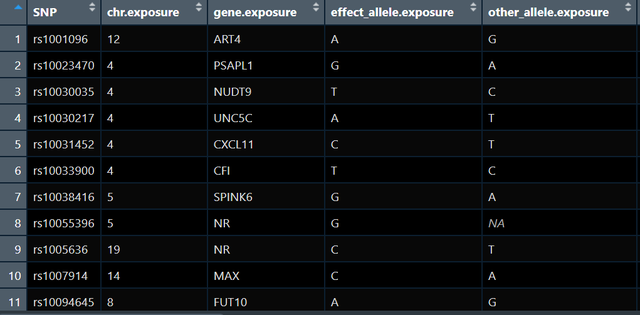
可以看到有很多个基因工具变量SNP,这个时候我们需要考虑连锁不平衡(linkage disequilibrium):
exposure_data<-clump_data(exposure_data, clump_r2 = 0.001)上面的代码中clump_r2则是设定的容许相关性,到这儿我们算是手动地将工具变量都筛出来了,解决了找工具变量的问题,还有一个方法是自动筛选工具变量,比如我暴露是bmi,我可以写出如下代码:
subset(ao, grepl("body mass", trait))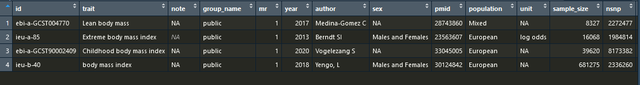
运行后我知道我可以选的gwasid是ieu-b-40,这个时候我也可以自动提取出工具变量,这两种方法达到的目的都是一样的:
extract_instruments('ieu-b-40')然后依照工具变量进行结局的summary estimates的提取,提取结局的summary data也需要是需要知道GWASid的对吧,比如我现在关心的结局是收缩压,我就可以写出如下代码:
outcome_gwas <- subset(ao, grepl("Systolic", trait))运行后我就可以知道所有的和收缩压相关的gwasid了,我选一个最新的,比如我就选下面的2021年的:
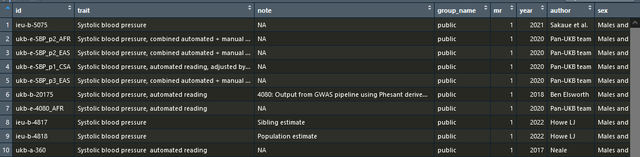
看图我们知道它对于的id是ieu-b-5075,我就这么写:
outcome_data <- extract_outcome_data(
snps = exposure_data$SNP, outcomes = "ieu-b-5075")后续通过合并直接做mr分析就可以,流程就没有不同了。
小结
今天给大家写了孟德尔随机话的实操,文章图示例来自【中文孟德尔随机化】英国布里斯托大学MRC-IEU《R语言做孟德尔随机化》第一章:用MRBase网页工具和R包TwoSampleMR做两样本孟德尔随机化_哔哩哔哩_bilibili,感谢大家耐心看完
R数据分析:孟德尔随机化实操的更多相关文章
- R数据分析:样本量计算的底层逻辑与实操,pwr包
样本量问题真的是好多人的老大难,是很多同学科研入门第一个拦路虎,今天给本科同学改大创标书又遇到这个问题,我想想不止是本科生对这个问题不会,很多同学从上研究生到最后脱离科研估计也没能把这个问题弄得很明白 ...
- Python相关分析—一个金融场景的案例实操
哲学告诉我们:世界是一个普遍联系的有机整体,现象之间客观上存在着某种有机联系,一种现象的发展变化,必然受与之关联的其他现象发展变化的制约与影响,在统计学中,这种依存关系可以分为相关关系和回归函数关系两 ...
- R数据分析:跟随top期刊手把手教你做一个临床预测模型
临床预测模型也是大家比较感兴趣的,今天就带着大家看一篇临床预测模型的文章,并且用一个例子给大家过一遍做法. 这篇文章来自护理领域顶级期刊的文章,文章名在下面 Ballesta-Castillejos ...
- R数据分析:潜类别轨迹模型LCTM的做法,实例解析
最近看了好多潜类别轨迹latent class trajectory models的文章,发现这个方法和我之前常用的横断面数据的潜类别和潜剖面分析完全不是一个东西,做纵向轨迹的正宗流派还是这个方法,当 ...
- R数据分析:临床预测模型中校准曲线和DCA曲线的意义与做法
之前给大家写过一个临床预测模型:R数据分析:跟随top期刊手把手教你做一个临床预测模型,里面其实都是比较基础的模型判别能力discrimination的一些指标,那么今天就再进一步,给大家分享一些和临 ...
- 号外号外:9月13号《Speed-BI云平台案例实操--十分钟做报表》开讲了
引言:如何快速分析纷繁复杂的数据?如何快速做出老板满意的报表?如何快速将Speed-BI云平台运用到实际场景中? 本课程将通过各行各业案例背景,将Speed-BI云平台运用到实际场景中 ...
- QVM 实操记 - 18.12.28
视频回放地址:https://i.iamlj.com/mp4/QVM-IMC-12.27-1080P.mp4 目录 目录 常规开发部署流程 准备工作 开发准备 网站部署 操作步骤 重装系统 LANP环 ...
- Python关于类的实操
实操一:总结 1.什么是绑定到对象的方法,如何定义,如何调用,给谁用?有什么特性? 2.什么是绑定到类的方法,如何定义,如何调用,给谁用?有什么特性? 3.什么是解除绑定的函数,如何定义,如何调用,给 ...
- Linux基础实操五
实操一:nginx服务 二进制安装nginx包1) 1)#yum clean all 2)#yum install epel-release -y 3)#yum install nginx -y 1) ...
随机推荐
- 创建deploymen的几种方式
创建deployment方式有两种,一种是命令直接创建,一种是使用yaml文件 1. 直接使用命令方式: --record 参数用来记录版本,也可以忽略,建议带上 kubectl create dep ...
- KingbaseES V8R6备份恢复案例之---同一数据库创建不同stanza备份
案例说明: 在生产环境,有的应用需要调用数据库的sys_rman做备份,为了区分数据库自身的sys_rman备份和应用的备份,可以使用不同的stanza name创建备份.本案例介绍了,如何在King ...
- KingbaseES V8R6C5 通过securecmdd工具手工脚本部署集群
案例说明: 对于KingbaseES V8R6C5版本在部集群时,需要建立kingbase.root用户在节点间的ssh互信,如果在生产环境禁用root用户ssh登录,则通过ssh部署会失败:V8R6 ...
- 这份数据安全自查checklist请拿好,帮你补齐安全短板的妙招全在里面!
企业数据安全自查Checklist! 快来对照表单,看看你的数据安全及格了吗? 一.京东云安全Checklist建议 京东云安全拥有业界领先的安全研究团队,经过多年实践与经验积累,京东云已面向不同业务 ...
- KMP&Z函数详解
KMP 一些简单的定义: 真前缀:不是整个字符串的前缀 真后缀:不是整个字符串的后缀 当然不可能这么简单的,来个重要的定义 前缀函数: 给定一个长度为\(n\)的字符串\(s\),其 \(前缀函数\) ...
- typora收费了,最后一个免费版提供下载
typora收费了,在这里,博主提供最后一个免费版下载,地址如下,顺便把typora导入和导出word时需要的工具也一同提供.最看不惯免费用着别人的软件,还搞引流的垃圾网站和公众号.地址如下 http ...
- centos系统安装MySQL8
使用yum仓库安装MySQL8 1.查看centos系统版本 # cat /etc/redhat-release CentOS Linux release 7.9.2009 (Core) 2.下载并安 ...
- CentOS7部署FastDFS+nginx模块
软件下载 # 已经事先把所需软件下载好并上传到/usr/local/src目录了 https://github.com/happyfish100/libfastcommon/archive/V1.0. ...
- K8S ingress控制器
文章转载自: K8S ingress控制器 (一)https://blog.51cto.com/u_13760351/2728917 K8S ingress控制器 (二)https://blog.51 ...
- Fluentd直接传输日志给MongoDB (standalone)
官方文档地址:https://docs.fluentd.org/output/mongo td-agent版本默认没有包含out_mongo插件,需要安装这个插件才能使用 使用的是td-agent,安 ...