GWAS:拒绝假阳性之case和control数量比例严重失衡的解决方案(SAIGE模型的应用)
一、为什么要校正case和control数量比例不平衡情况
试问作为生信届人员,最怕的是什么,当然是统计结果不靠谱。统计结果不靠谱包括两方面:一个是假阴性,一个是假阳性。假阴性可以理解为白天鹅被误当成丑小鸭了,假阳性可以理解为一大堆青蛙,你不知道哪个才是你的真命天子。假阴性就罢了,最多让你错过发现真理的机会,但万一假阳性呢,你拿着一个看似完美的结果吭哧吭哧做实验验证,一年半载的周期下来,什么结果都验证不出来,岂不是坑了做实验的人。因此,我们就要在源头上,把这个不靠谱的统计结果杜绝出去。
上一篇文章什么!GWAS研究中case和control的比例是有讲究的?就讲到GWAS分析中,如果case和control数量比例失衡的话,会产出非常多的假阳性结果,而用SAIGE模型做GWAS分析可以校正这种数量比例不平衡的情况。下面具体讲讲怎么应用SAIGE模型。
二、怎么校正:SAIGE的下载和安装
1、下载SAIGE
此操作在Linux上进行,系统要求R-3.5.1, gcc >= 5.5.0, cmake 3.8.1
wget https://github.com/weizhouUMICH/SAIGE/blob/master/SAIGE_0.35.8.1_R_x86_64-pc-linux-gnu.tar.gz
2、安装SAIGE
R CMD INSTALL SAIGE_XX_R_x86_64-pc-linux-gnu.tar.gz
3、安装SAIGE所依赖的其他R包:Rcpp, RcppArmadillo, RcppParallel, data.table, SPAtest, RcppEigen, Matrix, methods, optparse
以下两个方法二选一:
如果是用conda的话,则用以下命令:
conda install -n r-env r-Rcpp #r-env是指conda下的R环境
conda install -n r-env r-RcppArmadillo
conda install -n r-env r-RcppParallel
conda install -n r-env r-SPAtest
conda install -n r-env r-RcppEigen
conda install -n r-env r-optparse
也可以进入R,用install.packages( )安装:
install.packages("Rcpp")
install.packages("RcppArmadillo")
install.packages("RcppParallel")
install.packages("SPAtest")
install.packages("RcppEigen")
install.packages("optparse")
三、怎么校正:SAIGE的分析、解读
1、第一步,计算NULL GLMM
1)计算NULL GLMM的命令:
Rscript step1_fitNULLGLMM.R \
--plinkFile=./input/plinkforGRM_1000samples_10kMarkers \
--phenoFile=./input/pheno_1000samples.txt \
--phenoCol=y \
--covarColList=x1,x2 \
--sampleIDColinphenoFile=IID \
--traitType=binary \
--outputPrefix=./output/example \
--nThreads=4 \
--LOCO=TRUE
plinkFile为plink的输入文件(bed, bim, fam格式)
phenoFile文件格式如下,第一列代表研究的表型,第二列及第N-1列代表协变量,最后一列IID为个体的ID号:

--phenoCol=y # 指定你要研究的表型列名,在本次例子中,指定y的表型分析。
--covarColList=x1,x2 #指定加入的协变量
--sampleIDColinphenoFile=IID #指定样本的ID
--traitType=binary #指定研究的表型的类型,binary指二分类,即case和control
--outputPrefix #生成文件的输出路径
2)输出文件的结果解读:
这个步骤会生成三个文件,分别为:example.rda、example.varianceRatio.txt、example_30markers.SAIGE.results.txt
第一个文件:example.rda,是一个model file
可以用R打开:
load("./output/example.rda")
names(modglmm)
modglmm$theta
第二个文件:example_30markers.SAIGE.results.txt,随意选取位点的关联分析结果
第三个文件:example.varianceRatio.txt
2、计算每个marker的SPA得分
1)计算每个marker的SNP得分命令:
Rscript step2_SPAtests.R \
--dosageFile=./input/dosage_10markers.txt \
--dosageFileNrowSkip=1 \
--dosageFileNcolSkip=6 \
--dosageFilecolnamesSkip=CHR,SNP,CM,POS,EFFECT_ALLELE,ALT_ALLELE \
--minMAF=0.0001 \
--sampleFile=./input/sampleIDindosage.txt \
--GMMATmodelFile=./output/example.rda \
--varianceRatioFile=./output/example.varianceRatio.txt \
--SAIGEOutputFile=./output/example.plainDosage.SAIGE.txt \
--numLinesOutput=2 \
--IsOutputAFinCaseCtrl=TRUE
dosage_10markers.txt的文件格式如下,类似于plink的ped格式,前六列分别为:CHR, SNP, CM, POS, COUNTED, ALT, 后面为个体的ID号:

sampleIDindosage.txt文件为样本ID名:

example.rda、example.varianceRatio.txt为第一步生成的两个文件。
2)输出文件的结果解读:
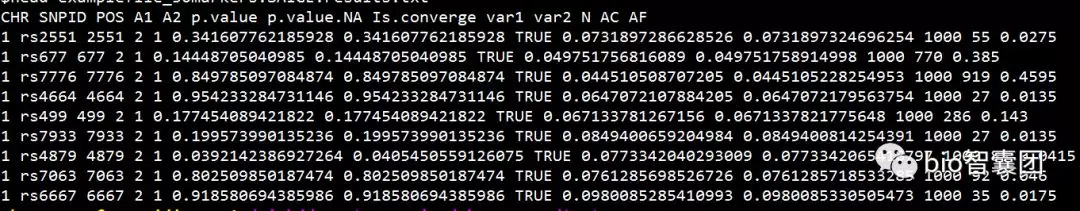
生成example.plainDosage.SAIGE.txt文件,其内容如下:

其中,P值(红框)即为我们校正case和control数量比例不平衡以后得到的GWAS结果,p.value.NA为不校正case和control数量不平衡的结果。
参数说明:
CHR: chromosome
POS: genome position
SNPID: variant ID
Allele1: Ref allele
Allele2: Alt allele
AC_Allele2: allele count of Alt allele
AF_Allele2: allele frequency of Alt allele
N: sample size
BETA: effect size
SE: standard error of BETA
Tstat: score statistic
p.value: p value with SPA applied
p.value.NA: p value when SPA is not applied
Is.SPA.converge: whether SPA is converged or not
varT: estimated variance of score statistic with sample related incorporated
varTstar: variance of score statistic without sample related incorporated
AF.Cases: allele frequency of allele 2 in cases (only for binary traits and if --IsOutputAFinCaseCtrl=TRUE)
AF.Controls: allele frequency of allele 2 in controls (only for binary traits and if --IsOutputAFinCaseCtrl=TRUE)
至此,校正GWAS分析中case和control数量比例严重失衡的工作就完成了。导师再也不用担心你给出一堆假阳性结果了。
GWAS:拒绝假阳性之case和control数量比例严重失衡的解决方案(SAIGE模型的应用)的更多相关文章
- GWAS研究中case和control的比例是有讲究的?
GWAS研究中,表型分两种.第一种是线性的表型,如果身高.体重.智力等:第二种是二元的表型,比如患病和未患病,即通常所说的case和control.对于表型是线性的样本来说,是不存在case和cont ...
- 在switch中的case语句中声明变量编译出错的解决方案
在switch中的case语句中声明变量编译的问题 先来看段代码,别管什么意思: : , j = ; ; i < ; i++) recive_phone[i] = msgbuf.text[i]; ...
- 全基因组关联分析学习资料(GWAS tutorial)
前言 很多人问我有没有关于全基因组关联分析(GWAS)原理的书籍或者文章推荐. 其实我个人觉得,做这个分析,先从跑流程开始,再去看原理. 为什么这么说呢,因为对于初学者来说,跑流程就像一个大黑洞,学习 ...
- GWAS | 全基因组关联分析 | Linkage disequilibrium (LD)连锁不平衡 | 曼哈顿图 Manhattan_plot | QQ_plot | haplotype phasing
现在GWAS已经属于比较古老的技术了,主要是碰到严重的瓶颈了,单纯的snp与表现的关联已经不够,需要具体的生物学解释,这些snp是如何具体导致疾病的发生的. 而且,大多数病找到的都不是个别显著的snp ...
- GWAS这十年 | 10 Years of GWAS Discovery: Biology, Function, and Translation
相关文章: A Unified Framework for Association Analysis with Multiple Related Phenotypes 太重要了,不得不单独拿出来分析一 ...
- 全基因组关联分析(Genome-Wide Association Study,GWAS)流程
全基因组关联分析流程: 一.准备plink文件 1.准备PED文件 PED文件有六列,六列内容如下: Family ID Individual ID Paternal ID Maternal ID S ...
- Oracle Applications Multiple Organizations Access Control for Custom Code
档 ID 420787.1 White Paper Oracle Applications Multiple Organizations Access Control for Custom Code ...
- if、else if 、else及switch...case使用小记(C#)
有时候编程编的久了,如果不停下来认真思考一下,即便是一些最基础的知识点,也可能让自己懵圈.其实,说到底还是打基础的时候没打牢,或者说自以为是地认为自己懂了,然后在打基础的时候就懒得思考懒得看了,结果就 ...
- JVM中可生成的最大Thread数量
最近想测试下Openfire下的最大并发数,需要开大量线程来模拟客户端.对于一个JVM实例到底能开多少个线程一直心存疑惑,所以打算实际测试下,简单google了把,找到影响线程数量的因素有下面几个: ...
随机推荐
- css 椭圆样式
<!DOCTYPE html> <html lang="en"> <head> <meta charset="UTF-8&quo ...
- vue中使用百度地图,悬浮窗搜索功能
https://www.cnblogs.com/shuaifing/p/8185311.html 侵删 <template> <div id="all"> ...
- Snowflake(雪花算法)的JavaScript实现
现在好多的ID都是服务器端生成的,当然JS也可以生成GUID或者UUID之类的,但是如果想要有序……这时就想到了雪花算法,但是都知道JS中Number的最大值为Number.MAX_SAFE_INTE ...
- 在VS 2017 下创建 Xamarin NuGet Package
最近在做一个Xamarin for android的项目,有个需求是一次可以从相册中选择多张图片,但是 android API<19 的版本还不支持一次选择多张图片,在网上找了一下,发现原生的组 ...
- 【设计模式】工厂方法模式 Factory Method Pattern
在简单工厂模式中产品的创建统一在工厂类的静态工厂方法中创建,体现了面形对象的封装性,客户程序不需要知道产品产生的细节,也体现了面向对象的单一职责原则(SRP),这样在产品很少的情况下使用起来还是很方便 ...
- 在Windows Phone 8.1中使用Sqlite数据库
前言 我的工作目前不涉及到Windows phone的开发,但是业余时间也开发过几款app.以前由于各种条件的限制,只接触到WP8.0设备的app开发. 最近几个月开始将WP8的应用迁移到WP8.1, ...
- java.lang.IllegalArgumentException: Called attach on a child which is not detached: ViewHolder
转载请标明出处,维权必究:https://www.cnblogs.com/tangZH/p/10116298.html 在项目过程中出现了上述错误. 会出现这样的错误是在我使用: notifyItem ...
- 【English】十、"谓语的地方"看到有两个动词:I go say hello.、非谓语形式
一.I go say hello. 这是一种偏口语的说法.一个句子中不能同时有两个谓语. 标准的用法有: I go and say hello. and 连接这两个动词,表示并列等关系.go and ...
- 【Oracle教程资源大合集】Oracle数据库免费学习资源汇总
Oracle的产品非常丰富,各类学习资源也五花八门,本文将介绍Oracle官方的免费教程与风哥整理的Oracle视频教程: 1.Oracle帮助中心 Oracle帮助中心也称为Oracle文档中心,这 ...
- SQLsever 复制一行内容到本表
insert into Table (userName,userAge) select userName,userAge from Table where Id=66 这里并不是 insert int ...