Pooled genome sequence strategies |representative genome assembly approaches|Domestication|GERP|selective sweep|Hybridization|Introgression|iHS|SNP genotyping arrays|haplotype
Design based on biology
通过比较基因组学的方法,将脊椎动物基因组的数据,解决生物学各方面问题。新的调控注释(在脊椎动物的进化过程中的出现的)可以丰富物种树(比如不同功能蛋白质进化速度上的差异(因为编码蛋白质基因和早期进化基因的发现))。
Sequencing 需要以下两种策略叠加:
1.Pooled genome sequence strategies :测同一物种的不同个体,不同个体叠加。
2.representative genome assembly approaches :因为有质量好的序列片段(reasonable N50 contig),所以可适用于缺乏长序列的情况。如果assembly质量好可以作为参考序列
Domestication:因为自然改变使得人改变,使得人为选择改变,人为改变部分导致的变化。
Project design:
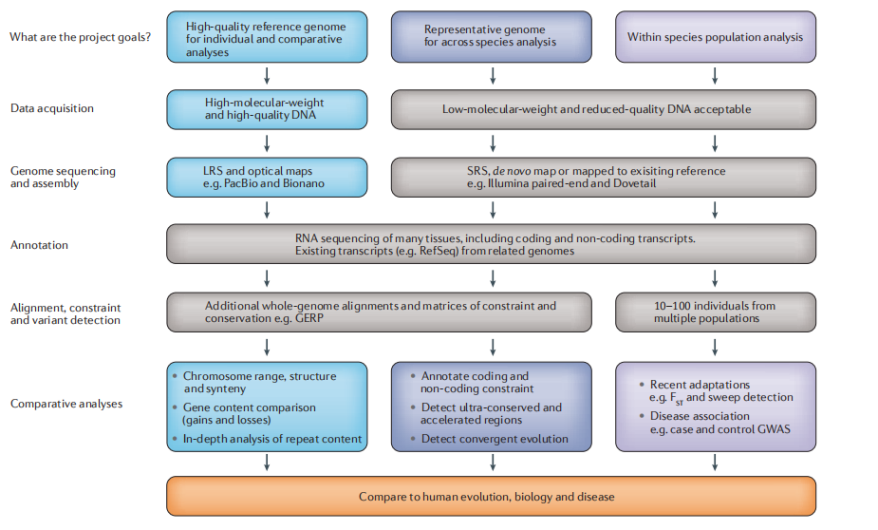

因为生物学分析依赖于assembly数据,所以assembly需要关注细节缩小误差(confounding effects)。
Data acquisition
Flowchart:1.什么样的测序方式对应什么样的研究2.除资源导向外,比较基因组学可以帮助找到内在机制3.
statistics:fixation index
GERP:genome evolutionary rate profiling:"GERP identifies constrained elements in multiple alignments by quantifying substitution deficits. These deficits represent substitutions that would have occurred if the element were neutral DNA, but did not occur because the element has been under functional constraint. We refer to these deficits as "Rejected Substitutions". Rejected substitutions are a natural measure of constraint that reflects the strength of past purifying selection on the element."
GWAS:genome-wide association studies
The effect of genome content:如果测序质量好,则可以扩大改数据的适用范围。测序质量的好坏与测序技术(以前的技术:radiation hybrids and BAC maps, BACs and fosmids,现在PacBio, Dovetail and Bionano)有关。
因为可以assembly大部分基因,所以邻近物种共线保守性可以用来研究基因组结构,新技术的综合使用使得assembly质量更好(N50变长),从而解决以前因技术不足造成的问题。由于脊椎动物基因组的复杂性(因为脊柱动物基因组有自己独特的特征:1.高重复2.高CG含量3.微染色体(质量较小的染色体)),更需要这种新技术(因为新技术对于重复区域可以单独span)。
Standing variation, imputation and mapping:
variation:发现突变重点在于选择样本:可以选择因为选择样本和探测差异同等重要,所以综合多个个体的低覆盖率序列(具体方式是综合使用软件探测差异)是合算的。
Imputation:确认发生突变的原因:可以通过1.计算遗传距离,2.滑动窗口model确认sweep或杂交或回交。
selective sweep:选择转移:因为对某位点的选择导致该位点周围的多样性下降。
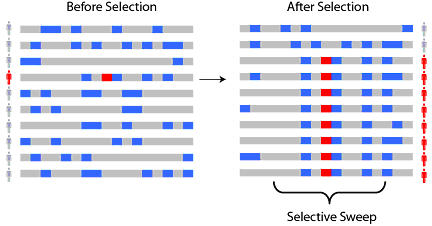
A selective sweep
Under natural selection, a new beneficial mutation will rise in frequency (prevalence) in a population. A schematic of polymorphisms along a chromosome, including the selected allele, before and after selection. Ancestral alleles are shown in gray and derived (non-ancestral) alleles are shown in blue. As a new positively selected allele (red) rises to high frequency, nearby linked alleles on the chromosome 'hitchhike' along with it to high frequency, creating a 'selective sweep.'
Hybridization:杂交:不同亲本之间杂交。
Introgression:回交:亲本和子代杂交。
Integrated haplotype homozygosity score:iHS (Integrated Haplotype Score) is a statistic that has been developed to detect evidence of recent positive selection at a locus. It is based on the differential levels of linkage disequilibrium(LD) surrounding a positively selected allele compared to the background allele at the same position.
Mapping:基因型与性状相对应:随着(1.SNP微阵列技术2.高通量测序价格下降),单倍体模型(通过足够的SNP密度数据)用于研究种群历史和基因型与性状的对应。
SNP genotyping arrays:SNP阵列是一种DNA微阵列,用于检测群体内的多态性。单核苷酸多态性是DNA中单个位点的变异,是基因组中最常见的变异类型。在人类基因组中已经鉴定了大约3.35亿个SNP,其中1500万个在全世界不同人群中以1%或更高的频率存在。
单倍型(haplotype:若干个决定同一性状的紧密连锁的基因构成的基因型
Complex mutation types: the good with the bad:
the bad:因为技术水平所限(SRS),所以许多高区域性突变(高区域性杂合和基因组断裂)无法找到,仅有少部分例子通过精确比对,可以解剖重排。
The good:现在出现了PacBio SMRT技术可以解开结构多样性
Layering complexity: gene and transcript annotation:
1.DNA Annotation>transcription annotation(方式1:比照相似物种的基因组;方式2:mapping RefSeq上的转录组)后可得到RNA序列(转录组,物种特异性的)
2.调节原件信息进行注释:调节原件导致突变(因为GWAS利用非编码区识别突变(GWAS可以map outside gene,以此达到检测疾病的目的。))
3.非编码区:GWAS基因组关联分析(genome-wide association studies, GWAS)已经被广泛用于复杂疾病的遗传位点的分析。 然而,GWAS 发现的复杂疾病相关的遗传变异,即单核苷酸多态性(SNP)位点大多位于基因的非编码区,并且同一区域中连锁的遗传变异(SNP)位点可以多达成百上千个。
4.特殊(特殊分类标准的)生物数据平台上的特殊data set进行注释
5.通过识别保守原件(来自不同物种的)进行注释
Vertebrate comparative genomics:Natural disease models: domestic animals:
物种分为模式生物和自然生物,自然生物正是研究稳态和健康特征的优选,所以在多目标的前体下驯化动物和自然动物都可以作为模型对象。
驯化的结果是表型的一致和疾病的富集。最近发现驯养动物有类人疾病(虽然没有实验室环境,但也是人类选择的结果)。用该动物不仅可以研究人类疾病,也可以造福该物种。
物种基因组比较,有助于annotation 2.通过GWAS找到SNP3.有助于找到sweep
Intraspecies comparison: a tool to study recent phenotypic adaptations:种内多个体比较可得到selective sweeps(特点是聚集多基因和基因多态)eg(不同季节的)鱼的单倍体基因长序列(coding区和非coding区,共同控制)控制一类及相关性状;eg不同海拔的sheep(由同一物种得到的性状different,采用不同物种作为验证。)
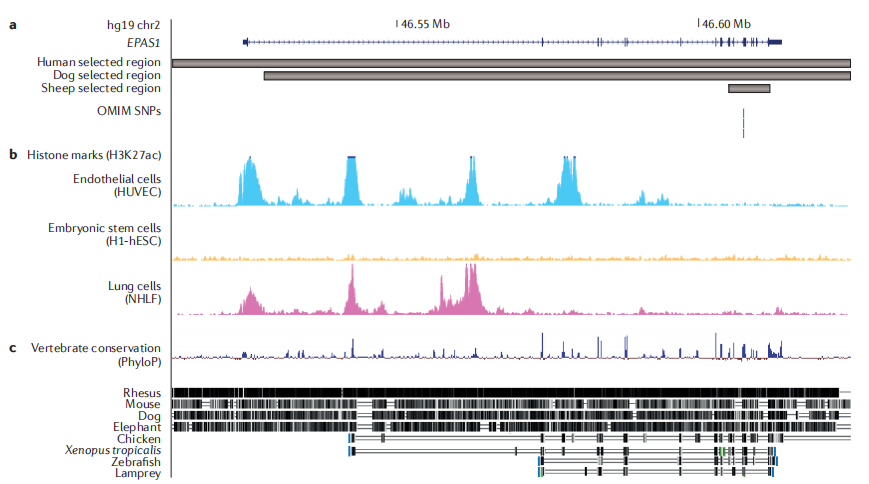


adaptations (microevolution)
Pooled genome sequence strategies |representative genome assembly approaches|Domestication|GERP|selective sweep|Hybridization|Introgression|iHS|SNP genotyping arrays|haplotype的更多相关文章
- The sequence and de novo assembly of the giant panda genome.ppt
sequencing:使用二代测序原因:高通量,短序列 不用长序列原因: 1.算法错误率高 2.长序列测序将嵌合体基因错误积累.嵌合体基因:通过重组由来源与功能不同的基因序列剪接而形成的杂合基因 se ...
- (转)8 reviews about de novo genome assembly
转自:http://dskernel.blogspot.com/2012/04/8-reviews-about-de-novo-genome-assembly.html 8 reviews about ...
- 短序列组装Sequence Assembly(转载)
转载:http://blog.sina.com.cn/s/blog_4af3f0d20100fq5i.html 短序列组装(Sequence assembly)几乎是近年来next-generatio ...
- 使用UCSC Genome Browser下载人类所有mRNA序列
打开UCSC Genome Browser官网.网址:http://genome.ucsc.edu/ 点击导航栏的Genome Data 在新的页面中,点击human,可快速定位至页面中人类基因组数据 ...
- Why you should QC your reads AND your assembly?
鲤鱼基因组:http://www.ntv.cn/a/20140923/52953.shtml 关于鲤鱼基因组的测定,数据质量控制遭到质疑. Why you should QC your reads ...
- 【bioinfo】生物信息学——代码遇见生物学的地方
注:从进入生信领域到现在,已经过去快8年了.生物信息学包含了我最喜欢的三门学科:生物学.计算机科学和数学.但是如果突然问起,什么是生物信息学,我还是无法给出一个让自己满意的答案.于是便有了这篇博客. ...
- GATK--使用转载
http://blog.sciencenet.cn/blog-1469385-819498.html 文章目录 一.准备工作 二.流程概览 三.流程 首先说说GATK可以做什么.它主要用于从seque ...
- GWAS Catalog数据库简介
GWAS Catalog The NHGRI-EBI Catalog of published genome-wide association studies EBI负责维护的一个收集已发表的GWAS ...
- GATK-BWA-MEM handle GRCh38 alternate contig mappings
1. For the Impatient # Download bwakit (or from <http://sourceforge.net/projects/bio-bwa/files/bw ...
随机推荐
- AP架构基本过程
①.有线网络搭建(vlan,dhcp,路由等.) ②.ap零配置启动,通过dhcp获取IP地址及网关ip,同时获取ac ip地址. ③.AP主动建立到达ac的capwap隧道. ④.ap与ac建议隧道 ...
- Java连载69-接受输入、用数组模拟栈
一.编写一个酒店管理系统 1.直接上代码 package com.bjpowernode.java_learning; public class D69_1_ { //编写一个程序模拟酒店的管理系 ...
- centos 7.4 安装docker 19.03.6 版本。附带离线安装包
说明: 1.此环境为未安装过docker服务的环境, 如果已经安装,则自行卸载. 2.以下环境中上传的包及离线yum源默认为/home目录下,如无特殊说明,以此目录为准 步骤一:下载docker离线安 ...
- PAT Advanced 1076 Forwards on Weibo (30) [图的遍历,BFS,DFS]
题目 Weibo is known as the Chinese version of Twitter. One user on Weibo may have many followers, and ...
- python数据拼接: pd.concat
1.concat concat函数是在pandas底下的方法,可以将数据根据不同的轴作简单的融合 pd.concat(objs, axis=0, join='outer', join_axes=Non ...
- python基础2--进制、字符编码和文件处理
一.进制 1.二进制 定义 二进制数据是用0和1两个数码来表示的数.它的基数为2,进位规则是"逢二进一" 转换方式 二进制转换为十进制: 把二进制数按权展开.相加即得十进制数. ...
- CentOS 7.3 下部署基于 Node.js的微信小程序商城
本文档为微信小程序商城NideShop项目的安装部署教程,欢迎star NideShop商城api服务:https://github.com/tumobi/nideshop NideShop微信小程序 ...
- java实现接口导出csv文件
Tomxin7 Simple, Interesting | 简单,有趣 业务介绍 项目要求从数据库中查询出相关数据后,通过表格展示给用户,如果用户需要,可以点击导出按钮,导出数据为csv格式. 开发环 ...
- 吴裕雄--天生自然C++语言学习笔记:C++简介
C++ 是一种中级语言,它是由 Bjarne Stroustrup 于 年在贝尔实验室开始设计开发的.C++ 进一步扩充和完善了 C 语言,是一种面向对象的程序设计语言.C++ 可运行于多种平台上,如 ...
- Okhttp 多次调用同一个方法出现错误java.net.SocketException: Socket closed
Okhttp 多次调用同一个方法出现错误java.net.SocketException: Socket closed https://blog.csdn.net/QQiqq1314/article/ ...