VCF和GVCF格式说明
注意:本文的内容主要来自于GATK官网的讲解,所以vcf也是GATK产生的,用其他caller,比如varscan2产生的vcf文件的内容注释可能不一致。
参考:https://gatkforums.broadinstitute.org/gatk/discussion/1268/what-is-a-vcf-and-how-should-i-interpret-it
VCF:由HEADER和RECORDS组成。
RECORDS的FORMAT内容详解:
QUAL:指的是caller正确的识别该变异位点的可能性,属于phred-scale quality score的一个应用。
GT,GQ,PL三者的关系:
GT是指该位点最有可能的基因型。
GQ是该位点第二有可能的基因型的PL值。
PL是不同基因型对应的标准化的可能性。
对于二倍体生物来说,PL有三个值,分别对应0/0,0/1,1/1。最有可能的基因型的PL值为0,第二小的是第二个可能。GQ反映的是第二个小的基因型的PL值,如果该值超过99,则定位99,因为超过了99,其几乎不能威胁第一个可能的地位。
计算方法:
PL(0/1) = 0 (the normalized value that corresponds to a likelihood of 1.0) as is always the case for the assigned allele,
but the next PL is PL(1/1) = 26 (which corresponds to 10^(-2.6), or 0.0025).
QUAL和GQ的区别:

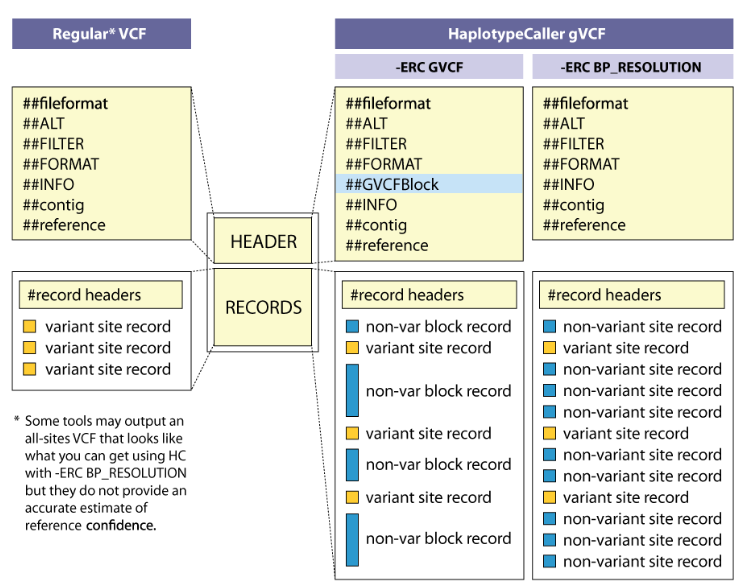
GVCF和VCF的最大区别是在于GVCF文件会记录所有的点,包括哪些没有突变的点。
在GVCF模式下,那些没有变异的点会形成一个未变异块,non-var block record。
GVCF的好处:能更方便把一群样本的GVCF联合起来,以便进行下一步分析,提高分析效率。而且GVCF的records还会提供一个
The records in a gVCF include an accurate estimation of how confident we are in the determination that the sites are homozygous-reference or not. This estimation is generated by the HaplotypeCaller's built-in reference model
VCF和GVCF格式说明的更多相关文章
- bcftools或vcftools提取指定区段的vcf文件(extract specified position )
下载安装bcftools 见如下命令: bcftools filter 1000Genomes.vcf.gz --regions 9:4700000-4800000 > 4700000-4800 ...
- GWAS | 全基因组关联分析 | Linkage disequilibrium (LD)连锁不平衡 | 曼哈顿图 Manhattan_plot | QQ_plot | haplotype phasing
现在GWAS已经属于比较古老的技术了,主要是碰到严重的瓶颈了,单纯的snp与表现的关联已经不够,需要具体的生物学解释,这些snp是如何具体导致疾病的发生的. 而且,大多数病找到的都不是个别显著的snp ...
- 收集vcftools所有用法
VCFtools用来处理VCF文档. 筛选特定突变 比较文件 总结突变 转化文件格式 验证并合并文件 取突变交集和差集 Get basic file statistics input可以为VCF或BC ...
- the pipeline of call SNP
######################################## ############### Mapping ################ ################## ...
- 【转】GATK使用方法详解(包含bwa使用)
一.使用GATK前须知事项: (1)对GATK的测试主要使用的是人类全基因组和外显子组的测序数据,而且全部是基于illumina数据格式,目前还没有提供其他格式文件(如Ion Torrent)或者实验 ...
- GWAS Catalog数据库简介
GWAS Catalog The NHGRI-EBI Catalog of published genome-wide association studies EBI负责维护的一个收集已发表的GWAS ...
- admixture 群体结构分析
tructure是与PCA.进化树相似的方法,就是利用分子标记的基因型信息对一组样本进行分类,分子标记可以是SNP.indel.SSR.相比于PCA,进化树,群体结构分析可明确各个群之间是否存在交流及 ...
- plink 进行PCA分析
当我们进行群体遗传分析时,得到vcf后,可利用plink进行主成分(PCA)分析: 一.软件安装 1 conda install plink 二.使用流程 第一步:将vcf转换为plink格式 1 p ...
- pysam - 多种格式基因组数据(sam/bam/vcf/bcf/cram/…)读写与处理模块(python)
在开发基因组相关流程或工具时,经常需要读取.处理和创建bam.vcf.bcf文件.目前已经有一些主流的处理此类格式文件的工具,如samtools.picard.vcftools.bcftools,但此 ...
随机推荐
- 【BZOJ3872】[Poi2014]Ant colony 树形DP+二分
[BZOJ3872][Poi2014]Ant colony Description 给定一棵有n个节点的树.在每个叶子节点,有g群蚂蚁要从外面进来,其中第i群有m[i]只蚂蚁.这些蚂蚁会相继进入树中, ...
- Python中的一些函数
1. 中文繁体/简体转换 下载 zh_wiki.py:https://github.com/skydark/nstools/blob/master/zhtools/zh_wiki.py 和 langc ...
- LeetCode 笔记系列一 Median of Two Sorted Arrays
题目:There are two sorted arrays A and B of size m and n respectively. Find the median of the two sort ...
- sql语句的安全性考虑
sql语句的应该考虑哪些安全性呢? 1.防止sql注入,对特殊字符进行转义(addslashes),或者使用已经编译好的sql语句进行变量的绑定: 2.当sql运行出现错误的时候,不要把数据库返回的错 ...
- PHP 支持8种基本的数据类型。
四种标量类型:boolean (布尔型):这是最简单的类型,只有两种取值,可以为 TRUE/true 或 FALSE/false ,不区分大小写.详细请查看:PHP布尔类型(boolean)integ ...
- 前端开发 - HTML
1.index2.head标签相关内容3.常用标签一4.常用标签二 table5.常用标签二 form6.标签分类 1.index <!--声明文档的类型 标记该文档为HTML5的文件--> ...
- SpringBoot与消息(RabbitMQ)
1. JMS和AMQP JMS(Java Message Service): ActiveMQ是JMS实现; AMQP(Advanced Message Queuing Protocol) 兼容JMS ...
- ssh登录服务器
ssh -i /home/zhangsuosheng/mykey.pub myusername@111.111.111.111
- 【numpy】
ndarray在某个维度上堆叠,np.stack() np.hstack() np.vstack() https://blog.csdn.net/csdn15698845876/article/det ...
- 如何实现手游app瘦身?
手游服务商来说,手游包体大一直是个很困扰的问题.一款手游产品而言,包体大小和更新方式对于有效用户的转化率往往起到非常关键的作用,话说手游安装包越小,用户转化率越高,那该如何实现app瘦身呢? 工具/原 ...