安装生物信息学软件-bowtie2
好吧,这是本周(2016.10.21-28)的学习任务之一:安装bowtie2并学习其使用方法&参数设置
所以,啃文档咯,官方文档Version 2.2.9 http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml
以下是我的整理。我不生产文档,我只是文档的搬运工么么哒~
Bowtie2适合将长度50-1000bp的reads比对到长的参考序列上。Bowtie 2 indexes the genome with an FM Index
(based on the Burrows-Wheeler Transform or BWT) 。输出结果为SAM格式。已集成在很多软件中,如
TopHat(a fast splice junction mapper for RNA-seq reads),
Cufflinks(transcriptome assembly and isoform quantitiation from RNA-seq reads),
Crossbow( cloud-enabled tool for analyzing reseuqncing data),
Myrna(a cloud-enabled tool for aligning RNA-seq reads and measuring differential gene expression)。
Bowtie1和2的区别:Bowtie 2's command-line arguments and genome index format are both different from Bowtie 1's.
1,bowtie1出现的早,所以对于测序长度在50bp以下的序列效果不错,而bowtie2在长度在50bp以上的更好。
2,Bowtie 2支持有空位的比对Number of gaps and gap lengths are not restricted, except by way of the configurable scoring scheme.
3,Bowtie 2支持局部比对(local, some chars will be omited/trimmed),也可以全局比对(end-to-end, all char participate)
4,Bowtie 2对最长序列没有要求,但是Bowtie 1最长不能超过1000bp。
5. Bowtie 2 allows alignments to [overlap ambiguous characters] (e.g. `N`s) in the reference. Bowtie 1 does not.
6,Bowtie 2不能比对colorspace reads.
7, Bowtie 2's paired-end alignment is more flexible. Try to find unpaired alignments for each mate。
8, Bowtie 2 reports a spectrum of mapping qualities, Bowtie 1 reports either 0 or high。
MUMmer: align 2 very large sequences(eg: 2 genomes)
NUCmer, BLAT/BLAST, Bowtie2: sensitive alignment to short ref seq(eg: a bacterial genome)
安装bowtie2: 直接下载bowtie2-2.2.9-linux-x86_64.zip,解压,修改环境路径即可
(设置环境路径请自行百度,或者参考这个http://www.cnblogs.com/pxy7896/p/5886305.html)
Scores: 更高分=更相似
--ma :match bonus
--mp :mismatch penalty
--np :penality for having N in either the read or the ref
--rdg :affine read gap penalty
--rfg :affine ref gap penalty
全局比对栗子:默认,高质量位点的mismatch罚分为-6,长度为2的gap罚分为-11(gap open-5, extension-3),如果在长度为50的read中只有这两个问题,则总分为-17。所以,最好的分数是0,指read和ref完全相同。
default min score threshold:
可以用--score-min设置
-0.6-0.6*L(read长度)
局部比对栗子:罚分同上,但每个match获得bonus,+2,则如果是上面情况,则得分为2*49-6-11=81
default min score threshold:
20+8*ln(L)(read长度)
paired-end read:两个mate的分数相加
Mapping quality:higher=more unique
如果一个read来自重复区域,则可能map到多个地方,所以需要知道比对的可信程度。在SAM文件中,MAPQ就是这个,值为Q=-10log10p。p是对比对错误的概率的估计。一个read越特殊,比对错误的概率越小,Q值越大。
bowtie2是对paired read的每个mate分别比对的,所以如果两个比对结果不符合预期(比如方向不合适或者距离不合适),就说是align discordantly,这种在研究structural variants时有用。
--ff --fr --rf: expected relative orientation of the mates
-I and -X: expected range of inter-mates distances
--no-discordant: 禁止找discordant alignments
--no-mixed: mixed mode指对一个pair找不到paired-end alignment时,为每个mate找unpaired alignments。关了这个会快一点点。
SAM 格式中有一些flag和optional fields描述了paired-end特征
Mates can overlap, contain, or dovetail each other
overlap:

contain: mate2刚好是mate1的子序列
dovetail: 咬合,延长

bowtie2默认overlap和contain是concordant的,dovetai是disconcordant的。所以:
--no-overlap和--no-contain使得这两种情况变为disconcordant
--dovetail: 让dovetail变为concordant
Reporting mode:
dafault mode: 找多个匹配,报告最好的
-D和-R: 会使程序变慢,但是增大了找到最好比对的可能性(针对有多个比对的)
-k mode: 找一个或多个匹配,全报告
-k N找最多N个匹配,按比对分数降序排序
-a mode: 找和报告所有的。对大基因组,这个会很慢。
bowtie2找匹配时采用随机的策略,会使用--seed产生随机数来选择需要报告的匹配。所以如果使用
--non-deterministic,则对同样的输入reads,可能会产生不同的比对结果输出。
To rapidly narrow the number of possible alignments that must be considered, Bowtie 2 begins by
extracting substrings ("seeds") from the read and its reverse complement and aligning them in an
ungapped fashion with the help of the FM Index.
-L: seed length
-i: interval between extracted seeds
-N: # mismatches permitted per seed
如果想要更sensitive的比对,可以减小L和i,增大N。同样,-D和-R也与此相关。
--n-ceil: upper limit on # N if valid
Presets: setting many settings at once
--very-sensitive: 等价于-D 20 -R 3 -N 0 -L 20 -i S,1,0.50 //可以查看文档得知
过滤:
bowtie2会在SAM记录里写出低质量read,YF:i SAM optional field 也会解释过滤它们的原因(多个原因只说一个),但是不会去比对它们。
YF:Z:LN read长度<=seed mismatches(-N)
YF:Z:NS read里N的数量>(--n-ceil)
YF:Z:SC 低于--score-min
YF:Z:QC 与--qv-filter设定相关。Illumina的QSEQ格式中,read最后一个域含1
small(32-bit numbers, for <4 billion nucleotides in length, index.bt2) and large(64-bit numbers, .bt21) index自动选择,无需担心
Performance tuning:
-p: 多线程
-o/--offrate: 使用bowtie2-build时,这个值比default设得小些。这样会有更大的index,适合-k和-a模式(报告多个比对的)。但如果计算机内存小,还是把这个值设大,减少内存消耗。
×××××××××××××××××××××××××××××××××××××××实验×××××××××××××××××××××××××××××××××××××××××××××××××××××××
Step 1:建索引
新建文件夹,在新文件夹下运行,
bowtie2-build /home/pxy7896/Downloads/bowtie2/example/reference/lambda_virus.fa lambda_virus
结果:产生六个文件。(eg1,eg2,eg3是后面的)
可以使用预先建好的索引。
可以一次为多个文件建立索引,文件名之间用,分隔
Step 2: 比对reads
bowtie2 -x lambda_virus -U /home/pxy7896/Downloads/bowtie2/example/reads/reads_1.fq -S eg1.sam
参数解释:
-x: 查看index文件首先在当前目录下找,找不到再去环境变量BOWTIE2_INDEXES下找。
-U: 后面跟需要比对的read文件。多个用,分隔。
-S: 后跟结果文件的名字。
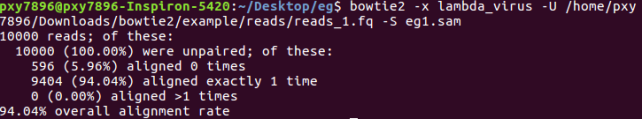
比对结果写到eg1.sam,要查看:head eg1.sam
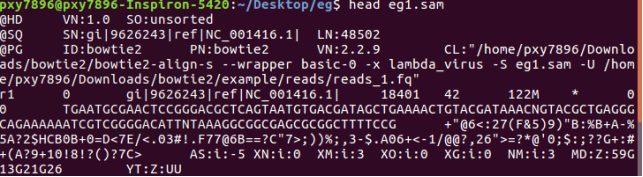
sam格式:(haven’t read yet)
Step 3: 比对paired-end reads
bowtie2 -x lambda_virus -1 /home/pxy7896/Downloads/bowtie2/example/reads/reads_1.fq -2 /home/pxy7896/Downloads/bowtie2/example/reads/reads_2.fq -S eg2.sam
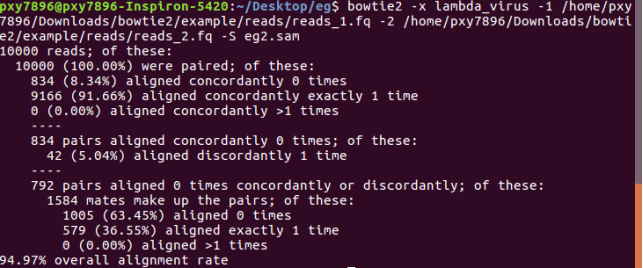
Step 4: long reads local alignment
bowtie2 --local -x lambda_virus -U /home/pxy7896/Downloads/bowtie2/example/reads/longreads.fq -S eg3.sam
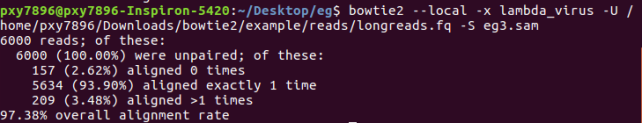
samtools:(可以输入命令,查询可以选择的参数)
Step 5: sam转为bam格式 (binary format of sam)
samtools view -b /home/pxy7896/Desktop/eg/eg2.sam -o eg2.bam
Step 6: sam排序为.sorted文件(这是压缩了的文件,适合存储)
samtools sort eg2.bam -o eg2.sorted
Step 7: generate variant calls in VCF format
发现居然没有装bcftools(for calling variants and manipulating VCF and BCF files)
sudo apt install bcftools
然后执行
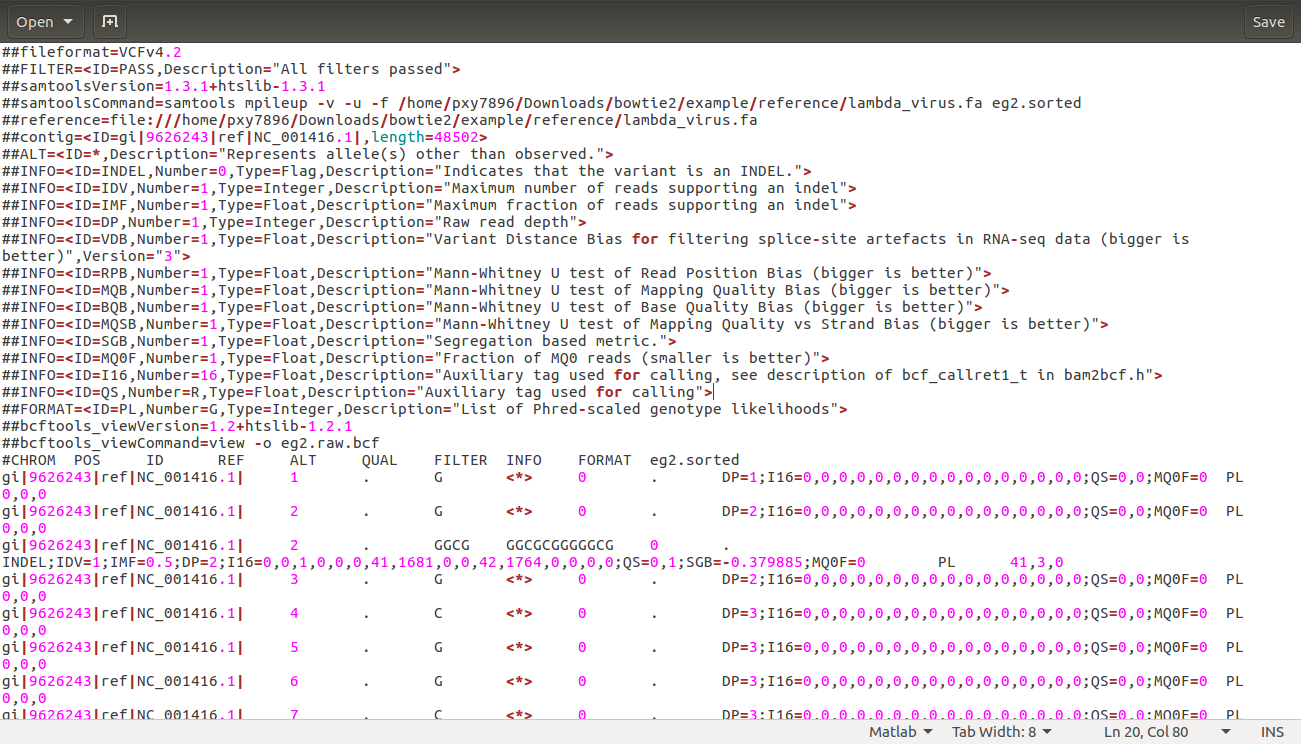
samtools mpileup -v -u -f $BT2_HOME/example/reference/lambda_virus.fa eg2.sorted | bcftools view -o eg2.raw.bcf
解释:将eg2.sorted和参考序列放一起比对,然后用bcftools将结果放到eg2.raw.bcf中
很奇怪,手册里的命令我运行的不对。。。可能还是这样找使用方法靠谱一点。
产生eg2.raw.bcf文件,大概长这个样子:
×××××××××××××××××××××××××××××××××××××××参数×××××××××××××××××××××××××××××××××××××××××××××××××××××××
bowtie2 
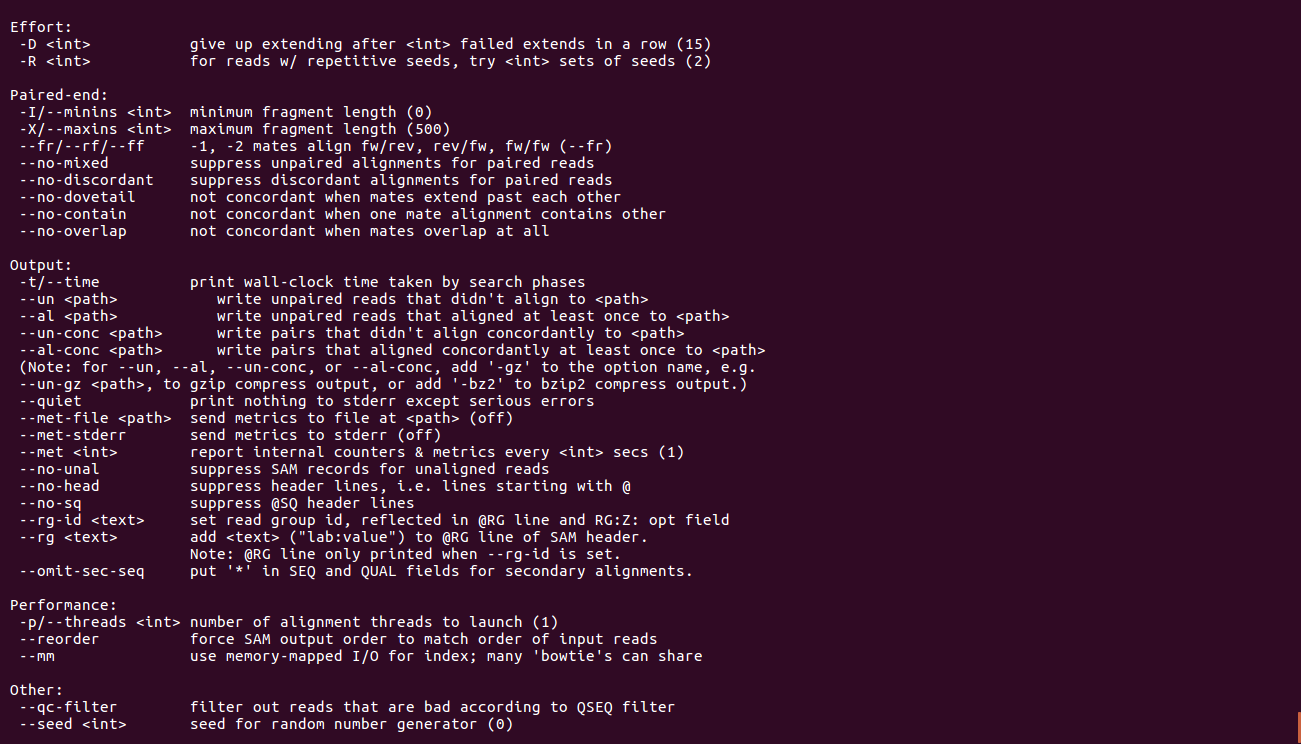

bowtie2-build(关于目录建立)
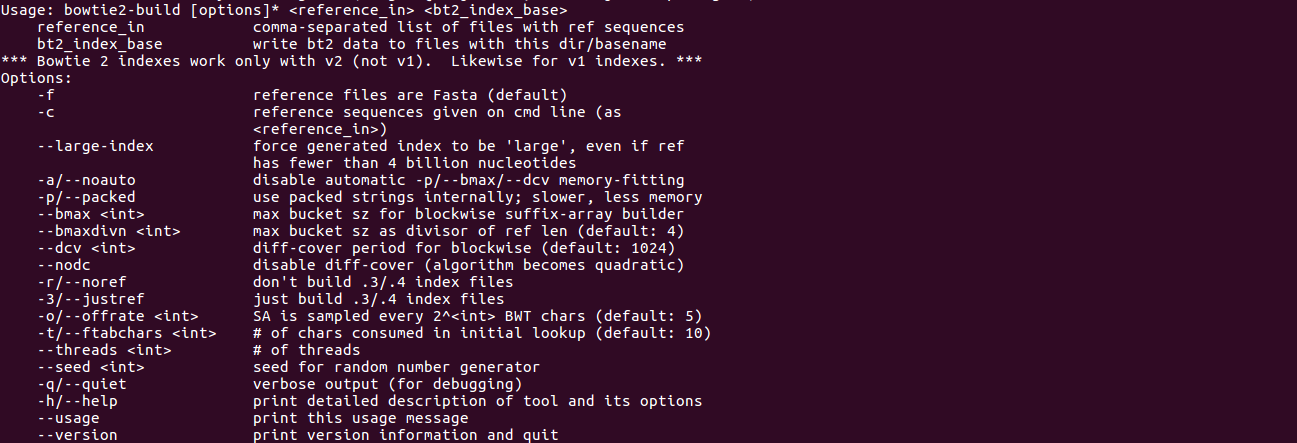
bowtie2-inspect
安装生物信息学软件-bowtie2的更多相关文章
- 安装生物信息学软件-MetaPhlAn2
上周20161021-20161028的任务还没有搞完,所以今天来填坑(微笑脸) ××××××××××××××××××××我是萌萌哒分割线××××××××××××××××××××××××××××××× ...
- 安装生物信息学软件-HUMAnN2
先挖坑 因为这个软件需要memory>16G,所以应该要安装在服务器上
- 安装生物信息学软件-Samtools
装完Bowtie2,官方文档给出的栗子说可以玩一玩samtools,所以我入个坑 参考这篇http://m.010lm.com/roll/2016/0620/2343389.html Step 1: ...
- 安装生物信息学软件-R
主要参考http://www.3fwork.com/b211/000091MYM021616/ Step 1 : sudo gedit /etc/apt/sources.list 添加到末尾 deb ...
- 安装生物信息学软件-Biopython
其实好多东西装过好多次,然而每次都要翻文档,经常掉进前面掉进过的坑...所以这里重新写一份指南,以防下次再装又忘了(魂淡我并不想再装了啊不要立flag) 1. 安装biopython 1.1 因为bi ...
- 单机静默安装GI软件并创建ASM实例和ASM磁盘组
环境:RHEL 6.4 + Oracle 11.2.0.4 需求:单机静默安装GI软件并创建ASM实例和ASM磁盘组,为后续迁移数据库文件到ASM做准备 1. 安装配置GI软件 2. 创建ASM实例 ...
- Linux删除apt-get安装的软件
我们都知道安装软件最简单的方法是apt-get install,但是卸载就不常用了,如何卸载呢?1.sudo apt-get remove xxxxxsudo apt-get autoremove2. ...
- Linux 中如何卸载已安装的软件(转载)
Linux 中如何卸载已安装的软件. Linux软件的安装和卸载一直是困扰许多新用户的难题.在Windows中,我们可以使用软件自带的安装卸载程序或在控制面板中的“添加/删除程序”来实 ...
- 安装ubuntu和安装ubuntu后要安装的软件列表
安装ubuntu 老毛桃进入win pe修复启动项 在win pe下面制作ubuntu的安装盘,只用的软件是ultraISO 参考博客下面的分区的那篇文章 http://jingyan.baidu. ...
随机推荐
- Android 图片的放大缩小拖拉
package com.example.ImageView; import android.annotation.SuppressLint; import android.content.Contex ...
- JS加密库Crypto-JS SEA加密
http://www.seacha.com/tools/aes.html 在该网站测试 CryptoJS有很多加密方式网上查阅后有 CryptoJS (crypto.js) 为 JavaScript ...
- Tiles & SiteMesh
Tiles & SiteMesh 这两天在给公司的新项目搭框架,在配tiles框架的时候发现一个小问题: 比如开发团队一共5人,每人10个页面,如果按照简单的tiles框架配置方法,每个 ...
- 。求推荐一个usb集线器的购买网址
笔记本蓝屏了,虽然后来让笔记本自己呆了好久,它冷静下来后我重新启动它,它又恢复了正常,但是我至今也没搞懂蓝屏的原因,深切地领悟到没文化不可怕,像我这样一知半解的最可怕... ------LYQ --- ...
- [poj1200]Crazy Search(hash)
Crazy Search Time Limit: 1000MS Memory Limit: 65536K Total Submissions: 26713 Accepted: 7449 Descrip ...
- python的ujson与simplejson
一.使用了simplejson import simplejson as json 二.使用ujson import ujson as json 参考链接:下载win下的:ujson
- C语言语法之关键字
注:该内容整理自以下链接. http://www.cnblogs.com/yezhenhan/archive/2011/10/16/2214420.html 由ANSI标准定义的C语言关键字共32个: ...
- C++ 不能在类体外指定关键字static
C++ static 函数的问题 近日读 C++ primer 中static 一章 , 有这么一句话, “静态成员函数的声明除了在类体中的函数声明前加上关键字static 以及不能声明为const ...
- Samba Linux 和windows 共享
1.安装Samba (yum install Samba) 2.配置Samba (Samba的配置文件为/etc/samba/smb.conf) 1)打开smb.conf vim /etc/s ...
- Find a point on a 'line' between two Vector3
Find a point on a 'line' between two Vector3http://forum.unity3d.com/threads/find-a-point-on-a-line- ...