Gromacs分子动力学模拟流程概述
Gromacs分子动力学模拟主要可以分为以下几个步骤,不同的体系步骤可能略有不同。
在开始之前,先简单了解一下预平衡:
分子动力学模拟的最终目的是对体系进行抽样,然后计算体系的能量,各种化学键,成分分析等等。打个比方说,我们有一个蛋白质,我们想将它放入一种溶液中(可能是水,也可能不是),然后看看这个体系的能量如何变化,蛋白质的化学键,与水分子形成的氢键等等信息,那么我们需要将蛋白质放入溶液中,映射到现实中就是讲溶剂放入溶剂中,然后等体系稳定后,观察其性质。
在MD中,这一过程不向现实中一样是自然发生的,我们需要通过模拟是体系演化到平衡状态,这就是预平衡。一般来说预平衡会有以下办法:
- 蛋白质结构能量最小化:PDB文件都是从晶体中获得的,所以蛋白质放入溶液中后必然会发生变化,这就需要对其进行能量最小化,确保蛋白质的结构是稳定结构。
- 蛋白质位置限定性模拟:有时加入溶剂后,分子间相互作用力会过大,导致蛋白质体系崩溃。这时我们需要限制蛋白质中重原子的位置,维持其结构,等溶剂分子弛豫之后再放开限制进行模拟。
- NVT预平衡,NPT预平衡:一般先做NVT模拟,减小盒子内压力,然后再做NPT模拟。
以上步骤当然不用全做,视情况而定,不过一般蛋白质能量最小化和位置限定性NPT还是要做的。
以下是分子动力学模拟的步骤,有些步骤可以省略。
- 获取并处理PDB文件
一般PDB文件是从网站上下载,如http://www.rcsb.org/pdb/home/home.do。获取PDB文件后有可能还要做一些处理,如末端氢原子,结晶水,等等。视情况而定。
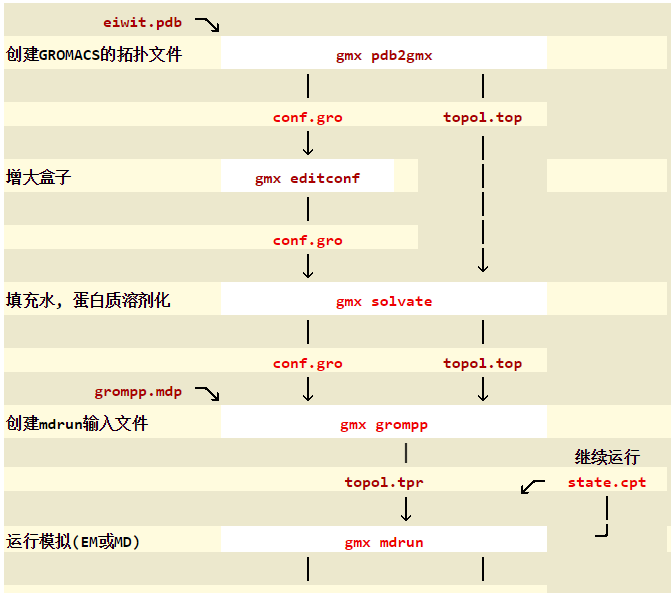
2. 使用pdb2gmx获得拓扑文件
命令pdb2gmx的详细信息可以参加http://manual.gromacs.org/programs/gmx-pdb2gmx.html。具体的命令参数我会在另一篇文章中详述。一般而言,我们使用时会是向下面这样:
gmx pdb2gmx -ff amber99sb-ildn -f *.pdb -o *.gro -p *.top -water tip3p
-ff 选项,制定要使用的力场;
-f选项,制定输入的PDB文件;
-o选项,制定生成的gro文件名
-p选项,制定要生成的拓扑文件名
-water选项,制定要使用的水分子模型
注意,除了生成*.gro文件和*.top文件之外,还会生成一个posre.itp,位置限定性文件(我把它理解成position-restraints的缩写)。
如果不使用-ff选项的话,指令运行后会让你自行选择力场。
3. 定义盒子
定义盒子和填充溶剂可以看做一步,在这里为了详细就分开来说。
与前面一样,涉及到的命令及文件都在其他文章中详述,下文不再赘述。使用editconf命令创建盒子:
gmx editconf -f *.gro -o *.gro -c -bt cubic -d 1.2
-f:指定输入的蛋白结构
-o:指定输出带盒子信息的结构文件
-c:将蛋白质置于盒子的中心,这个选项是可选的,不必须。
-d:蛋白质与模拟盒子在XYZ方向上的最小距离,一般不能小于0.9nm
-bt:指定盒子类型,这里使用了立方盒子,还可以用八面体,十二面体等。
这样我们就得到了周期型立方格子中的蛋白质分子。
editconf命令可以用于gro文件与pdb文件的相互转换。用-f指定源文件,-o指定所需文件名即可。
4. 蛋白质真空中的能量最小化(非必须)
一般而言这一步不是必须的,不过这里还是简述一下。如果我们只需要在真空中进行能量最小化的化,下一步就可以直接成品模拟了。
Gromacs使用grompp指令(GROMacs Pre-Preocessor)对带有格子信息的gro文件与蛋白质的拓扑文件,还有mdp文件进行处理,从而得到用于mdrun的输入文件*.tpr。tpr为二进制文件。具体指令如下:
gmx grompp -f *.mdp -c *.gro -p *.top -o *.tpr
-f:指定输入参数文件。mdp文件会有专门的文章叙述
-c:指定输入结构文件
-p:指定输入拓扑文件
-o:指定用于mdrun的tpr文件
运行之后我们得到*.tpr文件和参数文件mdout.mdp
然后使用mdrun命令运行能量最小化:
gmx mdrun -v -deffnm *
-v:显示模拟过程中的信息
-deffnm:我把它理解成define-file-name的缩写。定义输出文件名,文件后缀会自动加上。
运行后得到日志文件*.log,全精度轨迹问价*.trr,能量文件*.edr,能量最小化的结构文件*.gro。
5. 向盒子中填充溶剂
其实这只是一小步,同上,为了详细我把它单独列为一步。
使用solvate命令填充溶剂,以水为例:
gmx solvate -cp *.gro -cs *.gro -o *.gro -p *.top
-cp:指定需要填充水分子的体系,即前面我们用editconf得到的带格子的结构文件
-cs:指定要使用的水模型
-p:指定体系的拓扑文件(原蛋白质的拓扑文件),这样solvate就可以修改体系的拓扑文件。
-o:指定填充水分子后的输出文件
运行之后我们可以得到得到-o所指定的文件,并且-p指定的top文件也会发生改变。
6. 添加离子
向盒子中添加溶剂之后,我们得到了一个带电荷的溶液体系,因此必须进行中和。GROMACS中添加离子的指令是genion(我把它理解成generate-ion的缩写),但是不巧的是genion需要的输入文件为tpr文件。跟前面一样,这需要grompp(GROMacs Pre-Processor)来产生。grompp可以处理坐标文件和拓扑(描述分子的文件)从而产生原子级别的输入文件,即tpr文件,tpr文件包含了体系中所有原子的参数。
为了将坐标信息(gro)和拓扑信息(top)结合起来,我们需要一个mdp文件。mdp文件通常用于进行能量最小化,这里只是简单的生成tpr文件。
gmx grompp -f *.mdp -c *.gro -p *.top -o *.tpr
-f:指定mdp文件
-c:指定结构文件(加入溶剂后的结构文件)
-p:指定拓扑文件(还是之前生成的蛋白质拓扑,当然在加入溶剂时该文件发生了变化)
-o:指定输出文件
得到tpr文件后,就可以在其中加入离子了:
gmx genion -s *.tpr -o *.gro -p *.top - pname * -nname * -nn *
-s:将上述生成的tpr文件作为输入
-o:生成新的结构文件
-p:再次改变top文件,反应蛋白质结构的改变
-pname:指定要添加的阳离子名称,后面未指定数量,即为不添加
-nname:指定添加的阴离子名称
-nn:添加的阴离子数目
7. 能量最小化
现在,我们定义了盒子(周期性边界条件),溶剂分子,离子。整个体系已经到达电中性。在进行模拟之前,我们必须确保体系的结构正常,原子间距离不要太近,结合构型合理。这就需要对结构进行弛豫,这一过程称之为能量最小化(EM,energy minimization),是MD中非常重要的一步。
与前面类似,依然是需要用grompp来产生tpr文件,首先要定义一个minim.mdp文件,定义好之后:
gmx grompp -f minim.mdp -c *.gro -p *.top -o *.tpr
-f:指定mdp文件
-c:指定结构文件
-p:指定拓扑文件
-o:指定输出的文件名
得到tpr文件后就可以进行能量最小化了
gmx mdrun -v -deffnm em
mdrun的指令与前面一样。
我们将得到以下文件:
*.log 日志文件,记录了能量最小化过程
*.edr 二进制能量文件
*.trr 全精度的二进制轨迹文件
*.gro 能量最小化的结构
现在,我们的体系已经处于能量最小点了,可以做一些真正的模拟了!
8. NVT平衡
NVT平衡实际上是很重要的一步,但是它的核心在于mdp文件,而mdp文件我将在另一篇文章中单独阐述,因此这里对于NVT模拟就简化处理。
在一开始的pdb2gmx中我们生成了一个posre.itp 文件,这里终于派上用场了!它的作用是对蛋白质中的重原子(非氢原子)施加位置限制力。施加限制之后,这些原子就不能随便移动,除非能量非常大。这样做的目的在于平衡蛋白质周围分子的同时而不引起蛋白质结构的变化。
定义好mdp文件后,就可以进行模拟了。
gmx grompp -f *.mdp -c *.gro -p *.top -o *.tpr
-c指定前面生成的能量最小化的结构文件,-p依然指向那个被修改了多次的蛋白质top文件,-o指定输出文件。
gmx mdrun -deffnm *
指定输出文件名。
9. NPT平衡
与NVT平衡类似,关键在于mdp文件中,因此不再赘述,命令如下
gmx grompp -f *.mdp -c *.gro -t *.cpt -p *.top -o *.tpr
gmx mdrun -deffnm *
cpt为断点文件(check point),详见关于文件的文章中。
10. 成品MD
现在我们的体系已经在需要的温度和压强下平衡(弛豫)好了,我们可以放开位置限制并进行最终的MD,以收集数据了。
同样,先定义mdp文件,然后运行
gmx grompp -f *.mdp -c npt.gro -t npt.cpt -p *.top -o *.tpr
gmx mdrun -deffnm *
11. 分析
暂略,做到这里再补充。
Gromacs分子动力学模拟流程概述的更多相关文章
- 分子动力学模拟之基于自动微分的LINCS约束
技术背景 在分子动力学模拟的过程中,考虑到运动过程实际上是遵守牛顿第二定律的.而牛顿第二定律告诉我们,粒子的动力学过程仅跟受到的力场有关系,但是在模拟的过程中,有一些参量我们是不希望他们被更新或者改变 ...
- 分子动力学模拟之SETTLE约束算法
技术背景 在上一篇文章中,我们讨论了在分子动力学里面使用LINCS约束算法及其在具备自动微分能力的Jax框架下的代码实现.约束算法,在分子动力学模拟的过程中时常会使用到,用于固定一些既定的成键关系.例 ...
- Java虚拟机JVM学习01 流程概述
Java虚拟机JVM学习01 流程概述 Java虚拟机与程序的生命周期 一个运行时的Java虚拟机(JVM)负责运行一个Java程序. 当启动一个Java程序时,一个虚拟机实例诞生:当程序关闭退出,这 ...
- ETL流程概述及常用实现方法
ETL流程概述及常用实现方法 http://blog.csdn.net/btkuangxp/article/details/48224187 目录(?)[-] 1抽取作业 1手工开发抽取作业时候的常用 ...
- 分子动力学模拟软件VMD的安装与使用
技术背景 在分子动力学模拟过程中会遇到一些拓扑结构非常复杂的分子模型,所谓的复杂不仅仅是包含众多的原子,还有各种原子之间的成键关系与成键类型等.这时候就非常能够体现一个好的可视化软件的重要性了,这里我 ...
- MFCC特征参数提取流程概述
一 概念概述: 在语音识别(Speech Recognition)和话者识别(Speaker Recognition)方面,最常用到的语音特征就是梅尔倒谱系数(Mel-scale Frequenc ...
- 高复用率的RTSPClient组件EasyRTSPClient设计流程概述
EasyRTSPClient 设计过程 概述 EasyRTSPClient 基于live555构建而成. 今天讲讲EasyRTSPClient的设计过程 EasyRTSPClient,主要包括以下部分 ...
- Spark的任务提交和执行流程概述
1.概述 为了更好地理解调度,我们先看一下集群模式的Spark程序运行架构图,如上所示: 2.Spark中的基本概念 1.Application:表示你的程序 2.Driver:表示main函数,创建 ...
- Dubbo基础二之架构及处理流程概述
Dubbo基础一之实战初体验 - 池塘里洗澡的鸭子 - 博客园 (cnblogs.com)中体验了Dubbo的使用,对于消费端对服务提供者的调用非常清晰明确.那么Dubbo是如何做到的呢?下面对Dub ...
随机推荐
- OpenCV Java Tutorials- Camera Calibration
2020-10-10原文地址:https://opencv-java-tutorials.readthedocs.io/en/latest/09-camera-calibration.html#id1 ...
- win10简单方法安装杜比v4音效!win10 1909适用!
先下载这个! 链接: https://pan.baidu.com/s/1zAOOf-1aCJsjBgy36SiGWA 密码: s9n7 这个是杜比V4文件,257MB大小,适用32位64位系统!下 载 ...
- oh my zsh 常用插件
date: "2020-10-18T12:36:00+08:00" title: "oh my zsh 常用插件" tags: ["zsh" ...
- Zookeeper入门看这篇就够了
https://blog.csdn.net/java_66666/article/details/81015302
- jquery ui,拖拽,dragsort
一.导入js.导入jquery.dragsort.js外还需要导入jQuery. 二.HTML部分 <!DOCTYPE html> <html> <head> &l ...
- 2.通过QOpenGLWidget绘制三角形
参考:1.opengl绘制三角形 1.QOpenGLWidget的早先版本 QGLWidget是遗留Qt OpenGL模块的一部分,和其他QGL类一样,应该在新的应用程序中避免使用.相反,从Qt 5. ...
- Linux终端 terminal
终端 一个可以进行人机交互的界面 物理终端 设备终端:键盘.鼠标.显示器 终端类型 控制台终端: /dev/console 串行终端:/dev/ttyS# 虚拟终端:tty:teletypewrite ...
- 全球首个优秀的华人.net微服务框架 作者:百大僧
话不多说,直接上地址 https://gitee.com/shoubashou/NetCoreMicroService,目标斩获10000star, 通往牛逼的路上,风景差得让人只说脏话. 是全球首个 ...
- .net core autofac asyncinterceptor 异步拦截器帮助包
autofac使用拦截器实现AOP,是基于Castle.Core的.然而Castle.Core并未提供原生异步支持.所以需要使用帮助类实现,这在autofac官方文档的已知问题中有详细说明: http ...
- airtest操作夜神模拟器adb冲突解决办法
首先看错误日志明显是adb之间版本不对应互相干掉对方 第一步 去aritestIde所在目录\AirtestIDE\airtest\core\android\static\adb\windows 第二 ...