DNA序列编码中Hairpin的定义和计算
DNA序列编码中Hairpin的定义和计算
觉得有用的话,欢迎一起讨论相互学习~Follow Me
参考文献
[1] 张凯. DNA计算核酸编码优化及算法设计[D]. 2008.
[2] Shin, Soo Yong , et al. "Multiobjective evolutionary optimization of DNA sequences for reliable DNA computing." IEEE Transactions on Evolutionary Computation 9.2(2005):143-158.
[3] Shin, Soo Yong , I. H. Lee , and B. T. Zhang . "Evolutionary Multi-Objective Optimization for DNA Sequence Design." (2008).
[4] Shin, Soo Yong , et al. "Evolutionary sequence generation for reliable DNA computing." Congress on Evolutionary Computation IEEE, 2002.
[5] 饶泽书. 基于多目标粒子群的DNA编码算法研究[D]. 2018.
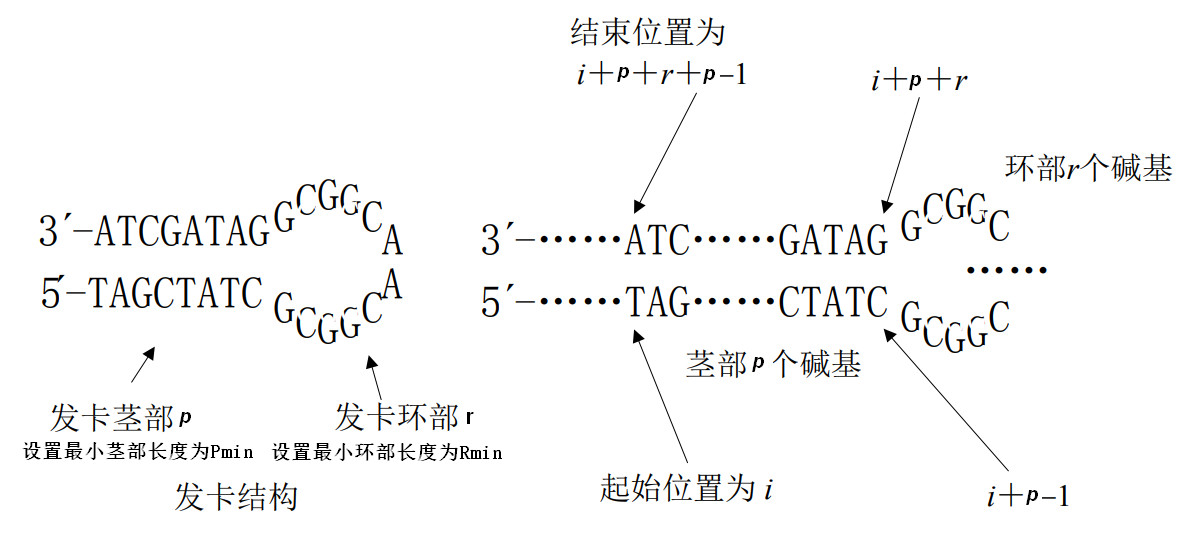
发卡结构约束
[ * ]定义
单链 DNA 分子产生二级结构通常由自身反向折叠而形成,发卡结构为典型的自身折叠结构.许多以特异性杂交反应为基础的 DNA 计算模型,都要求避免单链 DNA 形成二级
结构,这样单链 DNA 分子才能和自身的补链充分有效的发生特异性杂交[1]。
\[Hairpin( x ) = \sum _ { s = S _ { \mathrm { min } } } ^ { \left( l - R _ { \mathrm { min } } \right) / 2 } \sum _ { r = R _ { \mathrm { min } } } ^ { l - 2 s } \sum _ { i = 1 } ^ { l - 2 s - r } T \left( \sum _ { j = 1 } ^ { s } b p \left( x _ { s + i - j} , x _ { s + i + r + j-1} \right) , \frac { s } { 2 } \right)\]
- 式中s为茎长,Smin为设定的最小茎长。r为环长,Rmin为设定的最小环长,L表示DNA序列长度。在本文中,设置Smin=6,Rmin=6
- T表示阈值函数,T(x,y),只有在x>y时T(x,y)=x;否则T(x,y)=0,此处表示只有连续匹配达到了当前茎区数量的1/2(即>\(\frac{S}{2}\))才能算作为茎区的结构。
- bp(x,y)函数表示DNA序列中x和y位置的碱基相互互补的个数,如果相互互补即为1,否则记为0.
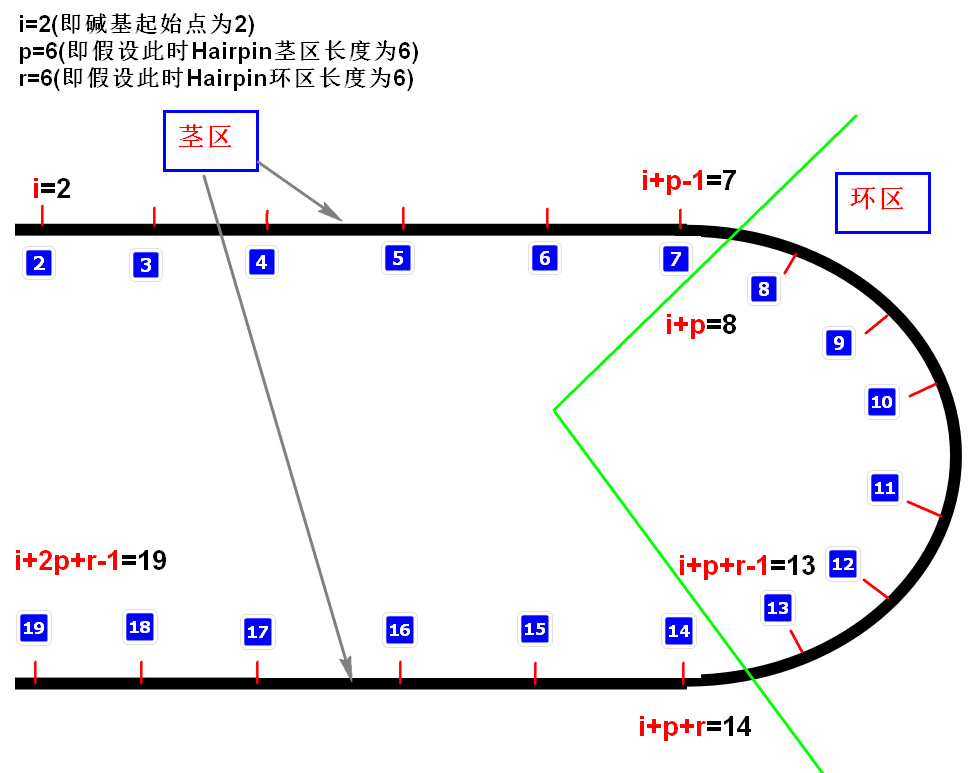
- s表示遍历茎区可能长度,其中 茎区最小长度为人为设定的Smin ,而 茎区最大长度是当环区长度取得最小值Rmin时的茎区长度(l-Rmin)/2
- r表示遍历环区可能长度,其中 环区最小长度为人为设定的Rmin ,而 **环区最大长度是当茎区长度取得最小值Smin时的环区长度l-2*Smin**
- i表示DNA序列起始处的索引,其中i最小从1处开始,最大可以到l-2s-r处,其中s和r皆为前两步中确定的值。
不同文章中发卡结构约束的定义及区别
上一章中定义此处标记为 [*]定义 而与其他定义相区别,其他定义则根据其引用的参考文献进行标记,即若此处定义出自于参考文献[1],则将其标记为 [1]定义
[2]定义

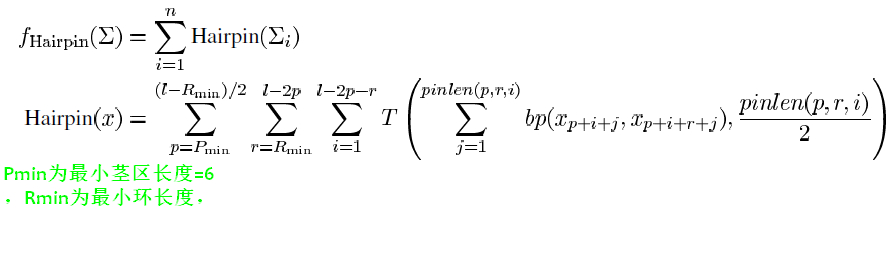
- 这个pinlen(p,r,i)很奇怪,定义为当假结中心在(p+i+r/2)时,可能的最大的茎区配对可能数 , 在作者2008发表的文章[3]中指出pinlen即为当前假定
的茎区数。 ==但是\(x_{p+i+j}和x_{p+i+r+j}的位置并不是假结茎区中一一对应的,[ * ]定义中是一一对应的关系\)==
[3]定义
- 在S.Y.Shin于2008年发表的[3]文章中,提出了如下定义:
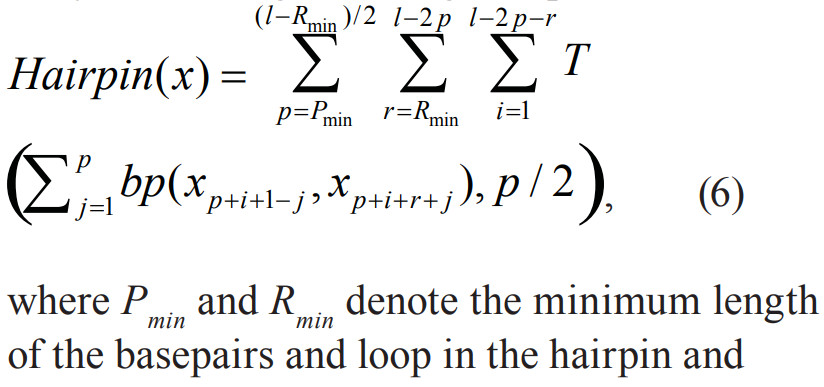
==[3] 定义与 [ * ]定义差别在于 [3] 定义中茎区匹配索引比 [ * ] 中均索引大1.==
[4]定义
- 在S.Y.Shin于2002年发表的[4]文章中,提出了如下定义:

其中Hairpin(x,c)函数没有明确的数学定义。仅仅是给出一个概念。
[5]定义
- \[\operatorname { Hairpin } ( x ) = \sum _ { s = S _ { \min } } ^ { \left( l - R _ { \mathrm { min } } \right) / 2 } \sum _ { r = R _ { \min } } ^ { l - 2 s } \sum _ { i = 1 } ^ { l - 2 s - r } T \left( \sum _ { j = 1 } ^ { s } b p \left( x _ { s + i - j } , x _ { s + i + r + j } \right) , \frac { S } { 2 } \right)\]
与[ * ]的区别在于\(x _ { s + i - j } , x _ { s + i + r + j }[5],x _ { s + i - j } , x _ { s + i + r + j-1 }[ * ]\)
分析与比较
可以看出[ * ]中Hairpin的计算公式较为正确
| No | J index | Expression x | Expression y |
|---|---|---|---|
| ==*== | - | \(x _ { s + i - j}\) | \(x _ { s + i + r + j-1}\) |
| - | j=1 | \(x_{s+i-1}\) | \(x_{s+i+r}\) |
| - | j=s | \(x _ {i}\) | \(x _ { i + r + 2s-1}\) |
| 2 | - | \(x_{s+i+j}\) | \(x_{s+i+r+j}\) |
| - | j=1 | \(x_{s+i+1}\) | \(x_{s+i+r+1}\) |
| - | j=s | \(x_{i+2s}\) | \(x_{i+r+2s}\) |
| 3 | - | \(x _ { s + i - j+1}\) | \(x _ { s + i + r + j}\) |
| - | j=1 | \(x_{s+i}\) | \(x_{s+i+r+1}\) |
| - | j=s | \(x _ {i+1}\) | \(x _ { i + r + 2s}\) |
| 5 | - | \(x _ { s + i - j}\) | \(x _ { s + i + r + j}\) |
| - | j=1 | \(x_{s+i-1}\) | \(x_{s+i+r=1}\) |
| - | j=s | \(x _ {i}\) | \(x _ { i + r + 2s}\) |
DNA序列编码中Hairpin的定义和计算的更多相关文章
- 在博客文章中使用mermaid 定义流程图,序列图,甘特图
概述 Mermaid(美人鱼)是一套markdown语法规范,用来在markdown文档中定义图形,包括流程图.序列图.甘特图等等. 它的官方网站是 https://mermaid-js.github ...
- [LeetCode] Repeated DNA Sequences 求重复的DNA序列
All DNA is composed of a series of nucleotides abbreviated as A, C, G, and T, for example: "ACG ...
- 华为OJ平台——DNA序列
题目描述: 一个DNA序列由A/C/G/T四个字母的排列组合组成.G和C的比例(定义为GC-Ratio)是序列中G和C两个字母的总的出现次数除以总的字母数目(也就是序列长度).在基因工程中,这个比例非 ...
- DNA序列对齐问题
问题描述: 该问题在算法导论中引申自求解两个DNA序列相似度的问题. 可以从很多角度定义两个DNA序列的相似度,其中有一种定义方法就是通过序列对齐的方式来定义其相似度. 给定两个DNA序列A和B,对齐 ...
- [DeeplearningAI笔记]序列模型1.1-1.2序列模型及其数学符号定义
5.1循环序列模型 觉得有用的话,欢迎一起讨论相互学习~Follow Me 1.1什么是序列模型 在进行语音识别时,给定了一个输入音频片段X,并要求输出片段对应的文字记录Y,这个例子中的输入和输出都输 ...
- python实现DNA序列字符串转换,互补链,反向链,反向互补链
在生物信息学分析中,经常对DNA序列进行一系列操作,包括子序列截取,互补序列获取,反向序列获取,反向互补序列获取.在python语言中,可编写如下函数完成这些简单功能. 子序列截取 python中对序 ...
- Leetcode 187.重复的DNA序列
重复的DNA序列 所有 DNA 由一系列缩写为 A,C,G 和 T 的核苷酸组成,例如:"ACGAATTCCG".在研究 DNA 时,识别 DNA 中的重复序列有时会对研究非常有帮 ...
- [LeetCode] 187. Repeated DNA Sequences 求重复的DNA序列
All DNA is composed of a series of nucleotides abbreviated as A, C, G, and T, for example: "ACG ...
- 利用Python【Orange】结合DNA序列进行人种预测
http://blog.csdn.net/jj12345jj198999/article/details/8951120 coursera上 web intelligence and big data ...
随机推荐
- 快速定位iOS线上BUG在哪个控制器崩溃
快速定位iOS线上App崩溃在哪个控制器里面,需要和后台配合使用 下载本SDK并手动添加到项目里 新建所有的页面都继承于YZViewController 在AppDelegate的didFinishL ...
- 【原】python3.7 无法pip安装提示ssl错误解决方案
问题 pip is configured with locations that require TLS/SSL, however the ssl module in Python is not av ...
- 【GAN与NLP】GAN的原理 —— 与VAE对比及JS散度出发
0. introduction GAN模型最早由Ian Goodfellow et al于2014年提出,之后主要用于signal processing和natural document proces ...
- 微软职位内部推荐-SW Engineer II for WinCE
微软近期Open的职位: Do you have a passion for embedded devices and services?   Does the following m ...
- mgo like的两种写法
实际上都是围绕正则来写的,看大家喜欢那种写法 package main import ( "fmt" "labix.org/v2/mgo" "labi ...
- PowerTeam--Alpha阶段个人贡献分及转会人员
PowerTeam--Alpha阶段个人贡献分 我们的团队共有6人,总分300分. 经团队成员通过个人申请以及组内投票的方式,最终的等级评定如下面的等级评定矩阵所示: β1 β2 β3 γ1 γ2 ...
- 2-Twelfth Scrum Meeting20151212
任务安排 成员 今日完成 明日任务 闫昊 获取视频播放的进度 获取视频播放进度 唐彬 解决handler可能引起的内存泄露问题 阅读IOS代码+阅读上届网络核心代码 史烨轩 下载service开发 ...
- 作业六:分析Linux内核创建一个新进程的过程
分析Linux内核创建一个新进程的过程 进程描述符PCB----task_struct数据结构 操作系统:1.进程管理 2.内存管理 3 文件系统 一.新进程如何创建和修改task_struct数据结 ...
- 20135327郭皓--Linux内核分析第三周 构造一个简单的Linux系统MenuOS
Linux内核分析第三周 构造一个简单的Linux系统MenuOS 前提回顾 1.计算机是如何工作的三个法宝 1.存储程序计算机 2.函数调用堆栈 3.中断 2.操作系统的两把宝剑 中断上下文的切换 ...
- java入门--4110:圣诞老人的礼物-Santa Clau’s Gifts
学习了一下java的语法,就用poj上的题目做作练习,好更快的熟悉常用的java语法. 题目在这里 http://bailian.openjudge.cn/practice/4110/ import ...