序列比对之Biostrings包
基本概念
Biostrings包很重要的3个功能是进行Pairwise sequence alignment 和Multiple sequence alignment及 Pattern finding in a sequence
序列比对一般有2个过程:
1)构建计分矩阵公式(the scoring matrix formulation)
2)比对(alignment itself)
global alignment methods (全局比对):align every residue in the sequences ,例如Needleman-Wunsch algorithm.
local alignment technique(局部比对): align regions of high similarity in the sequences,例如Smith-Waterman algorithm
安装
if("Biostrings" %in% rownames(installed.packages()) == FALSE) {source("http://bioconductor.org/biocLite.R");biocLite("Biostrings")}
suppressMessages(library(Biostrings))
ls('package:Biostrings')
----------------Pairwise sequence alignment---------------
步骤:首先构建罚分规则,然后按照规则进行比对。用pairwiseAlignment()函数
举例1:核酸序列
(myScoringMat <- nucleotideSubstitutionMatrix(match = 1, mismatch = -1, baseOnly = TRUE))#构建罚分规则
gapOpen <- 2 #gap分为2
gapExtend <- 1 #延伸gap分为1 sequence1 <- "GAATTCGGCTA" #序列1
sequence2 <- "GATTACCTA" #序列2
myAlignment <- pairwiseAlignment(sequence1, sequence2,
substitutionMatrix = myScoringMat, gapOpening = gapOpen,
gapExtension = gapExtend, type="global", scoreOnly = FALSE) #进行比对
myAlignment

举例2:对蛋白序列进行比对
蛋白比对会更复杂,因此模型更多,
data(package="Biostrings") #查看所有数据集
data(BLOSUM62) #这里选择BLOSUM62数据
subMat <- "BLOSUM62" #赋值
gapOpen <- 2
gapExtend <- 1
sequence1 <- "PAWHEAE"
sequence2 <- "HEAGAWGHE"
myAlignProt <- pairwiseAlignment(sequence1, sequence2,
substitutionMatrix = subMat, gapOpening = gapOpen, gapExtension =
gapExtend, type="global", scoreOnly = FALSE) #全局比对
myAlignProt2 <- pairwiseAlignment(sequence1, sequence2,
substitutionMatrix = subMat, gapOpening = gapOpen, gapExtension =
gapExtend, type="local",scoreOnly = FALSE) ##局部比对
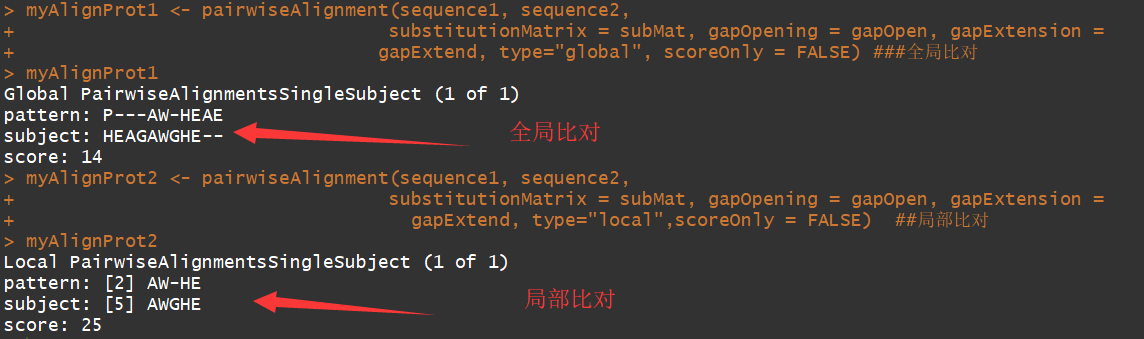
可以看到局部比对返回的是,高度相似的序列部分.
3)可视化,对于序列可以用最经典的对角线来可视化(以人和黑猩猩的hemoglobin beta为例)
library(seqinr) # 为了读取fasta序列
myseq <- read.fasta(file = "F:/R/Bioconductor/biostrings/prtein_example_seq.fas")
dotPlot(myseq[[1]], myseq[[2]], col=c("white", "red"), xlab="Human", ylab="Chimpanzee")

##########Multiple sequence alignment############
一般多序列比对可以用于进化分析
install.packages("muscle") #需要安装该包,因为该包在我的版本上没法安装,所以这里就不讲了
library(muscle)
######Phylogenetic analysis and tree plotting########
这里先不做分析
#########blast格式的解析######
install.packages("RFLPtools",dependencies=TRUE)
library(RFLPtools)
data(BLASTdata) #先查看数据集了解一下相关数据格式情况
head(BLASTdata)
colnames(BLASTdata)
DIR <- system.file("extdata", package = "RFLPtools") #用自带数据集
MyFile <- file.path(DIR, "BLASTexample.txt")
MyBLAST <- read.blast(file = MyFile)
mySimMat <- simMatrix(MyBLAST) #可以根据blast结果用来生成相似性矩阵,太厉害了
#########Pattern finding in a sequence######
library(Biostrings)
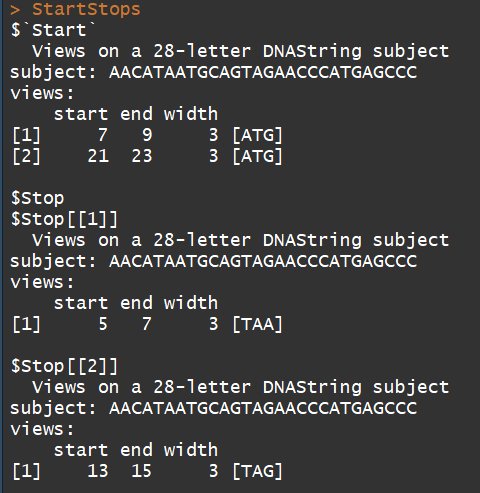
mynucleotide <- DNAString("aacataatgcagtagaacccatgagccc")
matchPattern(DNAString("ATG"), mynucleotide) #示例1
matchPattern("TAA", mynucleotide) #示例2
##以下函数可以用来寻找orf(需要修改)
myCodonFinder <- function(sequence){
startCodon = DNAString("ATG") # 指定起始密码子
stopCodons = list("TAA", "TAG", "TGA") # 指定终止密码子
codonPosition = list() #initialize the output to be returned as a list
codonPosition$Start = matchPattern(startCodon, sequence) # search start codons
x=list()
for(i in 1:3){ # iterate over all stop codons
x[[i]]= matchPattern(DNAString(stopCodons[[i]]), sequence)
codonPosition$Stop=x
}
return(codonPosition) # returns results
}
StartStops <- myCodonFinder(mynucleotide)
序列比对之Biostrings包的更多相关文章
- Python_序列与映射的解包操作
解包就是把序列或映射中每个元素单独提取出来,序列解包的一种简单用法就是把首个或前几个元素与后面几个元素分别提取出来,例如: first, seconde, *rest = sequence 如果seq ...
- Python 序列与映射的解包操作
解包就是把序列或映射中每个元素单独提取出来,序列解包的一种简单用法就是把首个或前几个元素与后面几个元素分别提取出来,例如: first, seconde, *rest = sequence 如果seq ...
- Python 序列与映射的解包操作-乾颐堂
解包就是把序列或映射中每个元素单独提取出来,序列解包的一种简单用法就是把首个或前几个元素与后面几个元素分别提取出来,例如: first, seconde, *rest = sequence 如果seq ...
- python基础之打/解包及运算符与控制流程
python基础之打/解包及运算符与控制流程 python中的解压缩(即序列类型的打包和解包) python提供了两个设计元祖和其他序列类型的处理的便利,也就是自动打包与自动解包功能,比如: data ...
- Oracle学习笔记一 初识Oracle
数据库简介 数据库(Database)是按照数据结构来组织.存储和管理数据的仓库.SQL 是 Structured Query Language(结构化查询语言)的首字母缩写词. 定义 数据库,简单来 ...
- Makefile <网络转载>
陈皓 (CSDN)概述——什 么是makefile?或许很多Winodws的程序员都不知道这个东西,因为那些Windows的IDE都为你做了这个工作,但我觉得要作一个好的和 professional的 ...
- oracle 体系结构
oracle 体系结构 数据库的体系结构是指数据库的组成.工作过程与原理,以及数据在数据库中的组织与管理机制. 1. oracle工作原理: 1).在数据库服务器上启动Oracle实例:2).应用程序 ...
- Navicat for Oracle实现连接Oracle
不知道为什么,从一开始,我就不喜欢Oracle,名字好听,功能强大,但总感觉"高不可攀";或许是因为我觉得其他的数据库就可以解决数据问题,不太了解Oracle的优势:而且它长得也不 ...
- 三、oracle 体系结构
1.oracle内存由SGA+PGA所构成 2.oracle数据库体系结构数据库的体系结构是指数据库的组成.工作过程与原理,以及数据在数据库中的组织与管理机制. oracle工作原理: 1).在数据库 ...
随机推荐
- linux 查看文件夹大小 du -h --max-depth=1 ./
du:查询文件或文件夹的磁盘使用空间 如果当前目录下文件和文件夹很多,使用不带参数du的命令,可以循环列出所有文件和文件夹所使用的空间.这对查看究竟是那个地方过大是不利的,所以得指定深入目录的层数,参 ...
- jcFlexible.js的小Demo
;(function(win, lib) { var doc = win.document; var docEl = doc.documentElement; var metaEl = doc.que ...
- Eclipse引入jar包的三种方式
引用自 http://chenxu.wo.blog.163.com/blog/static/50239687201162310620634/ 使用Eclipse开发应用程序,少不了使用第三方jar包, ...
- sql distinct 去除重复的字段
居然已经有人写了 那我就直接复制其链接吧
- PHP CI框架如何去掉 sql 里的反引号
在使用CI框架的时候, 经常的Active Record 类,这时候会出现一个问题 使用Active Record 类组成的sql 中,为了防止sql注入,会自动的在表名,字段名 自动添加反引号 当然 ...
- 关于后台程序java报错问题解决
HTTP Status – Internal Server Error Type Exception Report Message Handler processing failed; nested ...
- 小峰servlet/jsp(5)jsp自定义标签
一.自定义标签helloworld: 二.自定义有属性的标签: HelloWorldTag.java:继承TagSupport: package com.java1234.tag; import ja ...
- 一些css杂项笔记
div[class*="col-"]{ background-color: gold; border: 1px solid #ccc; } //给class开头等于col-的div ...
- 数据读取速度达1.5G/s,UFS 2.1存储技术曝光
目前最快的是苹果NVME,当然UFS2.1也不差 iPhone6s与iPhone6s Plus在硬件的规格上有了很大的提升,但是它们身上的变化远没有苹果在发布会上所提到的A9处理器.1200万摄像头以 ...
- C#中char空值的几种表示方式
C#中char空值的几种表示方式 在C#中char类型的表示方式通常是用单引号作为分隔符,而字符串是用双引号作为分隔符. 例如: 程序代码 程序代码 char a = 'a'; char b = 'b ...