tophat-fusion 鉴定融合基因
tophat-fusion 是一款利用RNA_seq 数据鉴定融合基因的工具,官网链接如下:
http://ccb.jhu.edu/software/tophat/fusion_index.shtml
安装:
tophat-fusion 是集成在tophat软件中的,具体的安装参考tophat的安装就好了
使用方法:
对于tophat-fusion 而言,要求固定的目录结构,比如我在result 文件夹下进行tophat-fusion的分析
那么我需要在该目录下准备几个文件:
1)物种对应的refGene.txt 和 ensGene.txt (这两个文件可以从UCSC下载得到)
2) 新建一个blast 文件夹,注意文件夹的名字必须为"blast", 在blast 文件夹下需要从NCBI下载 nt. human_genomic. other_genomic 开头的所有文件
下载的链接如下:
3) tophat_fusion 的输出目录: 每个样本一个输出目录,输出目录的前缀为tophat_, 下划线之后加上样本名称,类似 tophat_MCF7;
当然你还需要物种对应的bowtie1 的索引文件,注意这里必须为bowtie1的索引,tophat 检测融合基因时推荐bowtie1的索引方式
上述文件都准备好之后,就可以开始分析了;
第一步:toohat 比对,和普通的比对类似,只不过为了融合基因的检测,需要添加几个额外的参数:
tophat2 -o tophat_MCF7 -p 20 --fusion-search --keep-fasta-order --bowtie1 --no-coverage-search -r 0 --mate-std-dev 80 --max-intron-length 100000 --fusion-min-dist 100000 --fusion-anchor-length 13 --fusion-ignore-chromosomes chrM hg19_bowtie1/hg19 SRR064286_1.fastq SRR064286_2.fastq
第二步:tophat-fusion-post , 生成融合基因的结果
tophat-fusion-post -p 20 --num-fusion-reads 1 --num-fusion-pairs 2 --num-fusion-both 5 hg19_bowtie1/hg19
需要指出的是,tophat-fusion-post 根据固定的目录结构进行样本,如果有多个样本,每个样本单独进行tophat 比对,只要输出目录区分开即可,比如A,B,C 3个样本,就有3个输出文件夹
tophat_A, tophat_B, tophat_C
运行完成之后,会生成一个名为 tophatfusion_out 的文件夹,该文件夹下是所有样本的融合基因分析的结果:
1)result.hml : 所有样本的融合基因分析的结果,直接看这个html
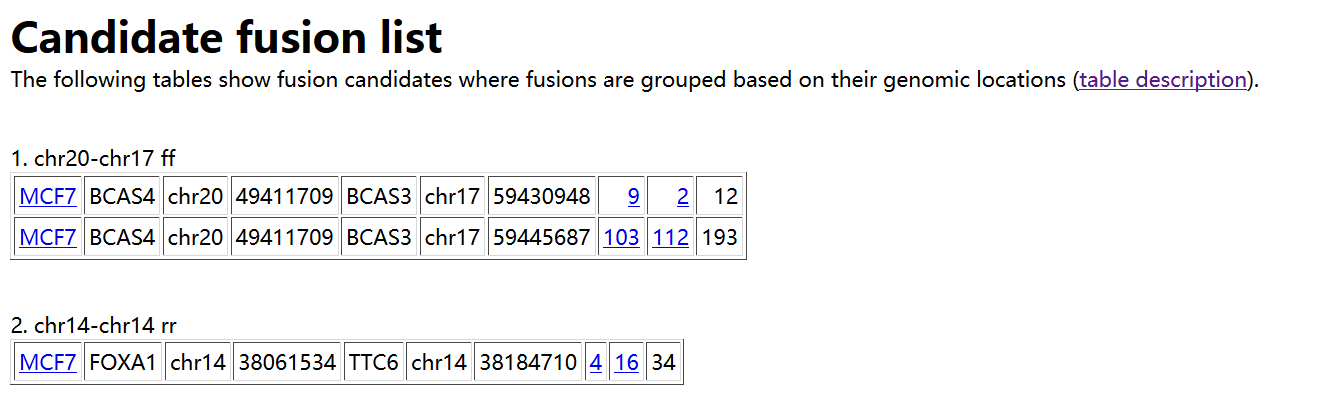
如上所示。在result.html 中,首先给出预测得到的融合基因,以表格形式进行展示,每列的含义如下:
1. Sample name in which a fusion is identified
2. Gene on the "left" side of the fusion
3. Chromosome ID on the left
4. Coordinates on the left
5. Gene on the "right" side
6. Chromosome ID on the right
7. Coordinates on the right
8. Number of spanning reads - this is the number of reads that span a
fusion point all on their own. In other words, the read itself has a
fusion break point within it.
9. Number of spanning mate pairs - this is the number of pairs of reads
where one read maps entirely on the left and the other read maps
entirely on the right of the fusion break point. Neither read is split,
so these pairs are not counted at all in (8).
10. Number of spanning mate pairs where one end spans a fusion (reads spanning fusion with only a few bases are included).
If you follow the the 9th column, it shows coordinates "number1:number2"
where one end is located at a distance of "number1" bases from the left
genomic coordinate of a fusion and "number2" is similarly defined.
tophat-fusion 鉴定融合基因的更多相关文章
- RNA-seq差异表达基因分析之TopHat篇
RNA-seq差异表达基因分析之TopHat篇 发表于2012 年 10 月 23 日 TopHat是基于Bowtie的将RNA-Seq数据mapping到参考基因组上,从而鉴定可变剪切(exon-e ...
- Mol Cell Proteomics. | 用于鉴定新型融合转录本及其在癌细胞中的潜在翻译产物的多功能蛋白质组基因组学工具FusionPro
期刊:Molecular & Cellular Proteomics 发表时间:June 17, 2019 DOI:10.1074/mcp.RA119.001456 分享人:任哲 内容与观点: ...
- 使用Tophat+cufflinks分析差异表达
使用Tophat+cufflinks分析差异表达 2017-06-15 19:09:43 522 0 0 使用TopHat+Cufflinks的流程图 序列的比对是RNA分析 ...
- FusionCancer-人类癌症相关的融合基因的数据库
RNA-seq 测序可以用于融合基因的发现,在过去的十几年里,RNA-seq 测序数据不断增加,发现的融合基因的数据也不断增加: FusionCancer 是一个人类癌症相关的融合基因的数据库,利用N ...
- FusionMap 检测融合基因
定义:融合基因是指两个或者多个基因联合起来,一起转录形成一个转录本: 检测的意义:融合基因可以作为某些疾病的特异分子标记,比如 bcr/abl融合基因存在于95%以上的慢性粒细胞白血病患者中: AML ...
- BCR-ABL融合基因及检测
费城染色体 费城染色体(Philadelphia chromosome, Ph (or Ph') chromosome),或称费城染色体易位(Philadelphia translocation),是 ...
- Mac入门 (二) 使用VMware Fusion虚拟机
有了Mac机,还是需在Mac上用Windows怎么办?, VMware Fusion 是运行在Mac机上的虚拟机软件, 类似于VMware workstation. 这样就可以在Mac上运行Windo ...
- Kinect for Windows SDK开发入门(十九):Kinect Fusion
Kinect for Windows SDK1.7中引入了Kinect Fusion功能.在1.8的SDK中对该功能进行了改进和强化,Kinect Fusion能够使得我们使用Kinect f ...
- 【记录】vmware fusion 7 windows 10 unidentified network
今天在 vmware fusion 7 中,使用 windows 10 时,突然报出一个错误(忘记截图了),当时就要求强制重启系统,也没怎么在意,但是重启之后,发现 windows 10 居然不能联网 ...
随机推荐
- 【驱动】DM9000A网卡驱动框架源码分析
Linux网络设备结构 首先看一下Linux网络设备的结构,如下图: 网络协议接口层向网络层协议提供提供统一的数据包收发接口,不论上层协议为ARP还是IP,都通过dev_queue_xmit()函数发 ...
- webpack的代码分割/离
两种方法: 1.webpack的methods----require.ensure 2.ES 2015的Loader spec //require.ensure语法 require.ensure [] ...
- CentOS6.5 安装Nexus配置Maven私服
1.下载Nexus的tar包,链接地址.注意,3.x版本需要JDK版本1.8及以上版本. 2.创建安装包存放目录 命令:mkdir -p /usr/local/src/installs 3.rz或者f ...
- 内部排序比较(Java版)
内部排序比较(Java版) 2017-06-21 目录 1 三种基本排序算法1.1 插入排序1.2 交换排序(冒泡)1.3 选择排序(简单)2 比较3 补充3.1 快速排序3.2 什么是桶排序3.3 ...
- libevent源码分析:evmap_io_active_函数
evmap_io_active_函数用于将激活指定文件描述符上的事件 void evmap_io_active_(struct event_base *base, evutil_socket_t fd ...
- 【转】31个实用的find命令
find . -name "*.sql" -exec md5sum {} \; 一.主要内容 ====================================== . 用文 ...
- Scala开发入门指南
作者:chszs,转载需注明.博客主页:http://blog.csdn.net/chszs 一.下载Scala 当前Scala的最新版本为2.10.2版,Windows有两种发布包: 1)Windo ...
- MAC层作用
对于无线传感网 MAC,顾名思义,就是介质访问控制,是用来控制无线介质的访问的,由于无线传输是共享空中资源的,必然存在多个无线传感器节点对传输介质的争用,MAC层协议就是用来解决这个问题的,包括冲突的 ...
- Gnome3 安装系统监视器
. . . . . 之前使用Gnome2的时候可以向面板上添加一个系统监视器,相信很多人都用过这个实用的小工具,可以很方便的了解系统的运行概况.但是自从使用了Gnome3之后这个小工具不见了,Gnom ...
- 阿里云RDS上用mysqldump导入导出
文章转载自: http://blog.csdn.net/jk0803_wantao/article/details/9179217 1. 开通云服务器服务.2. 开通RDS服务,如果开通成功,会返回给 ...