circRNA
一、circRNA序列提取
环状RNA (circRNA)是一类不具有 5' 末端帽子和 3' 末端 poly(A)尾巴、并以共价键形成环形结构的非编码 RNA 分子。 环状RNA (circRNA) 是区别于传统线性 RNA 的一类新型 RNA,大量存在于真核转录组中且表达具有时空特异性。在调控基因转录、作为疾病诊断marker等方面具有重要的研究和临床意义。
在预测circRNA时,都是检测breakpoint 处的reads 数,最后给出的环状RNA的ID 都是诸如 chr14:106994222-107183708 这样给出了起始和终止位置;对于某一个基因来说,其可能产生的circRNA的类型是多样的,以下图为例进行说明

1)由单个外显子组成的环状RNA, 比如
2)有多个外显子组成的环状RNA, 比如

以上的两种circRNA在序列提取时都非常容易,只需要将circRNA的起始和终止位置能够和某些外显子正好对应上,那么就可以确定其序列就是起始外显子和终止外显子之间的所有外显子构成的序列
3)只由内含子组成的环状RNA

这种环状RNA也可以方便的提取序列,直接确定起始和终止位置在基因组上的位置,将对应的序列提取出来即可
4)起始外显子和终止外显子之间有多个外显子,比如
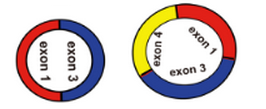
5)起始外显子和终止外显子之间有内含子,比如
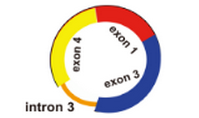
预测环状RNA时,只能够确定起始外显子和终止外显子,却不能确定在该circRNA中间到底有哪几个外显子,而且到底包不包含内含子序列,由于可变剪切的存在,可能存在多个外显子,也可能包含内含子,是不能够准确的提取circRNA对应的序列;能够做的只是将包括起始外显子和终止外显子以及之间的所有外显子连起来作为circRNA的序列
以上面的exon1-exon4 之间形成的环状RNA为例,我们只能将exon1-exon2-exon3-exon4的序列作为该环状RNA的序列,但是和实际的环状RNA的序列肯定是存在误差的;
目前分析手段没办法很好的解决这个问题,也许随着对环状RNA认识的加深和分析方法的改进,可以准确的识别circRNA的序列;为了准确的确定circRNA的序列,只能是针对breakpoint 两边的序列设计特异性引物,将circRNA 扩增出来,再测序,准确的识别序列;
二、提取fa
假设存在一个exon的bed文件,以及参考基因组的fa序列和gtf文件,如何根据reference和gtf文件,提取这个exon_bed文件中区间内的所有外显子序列。如exon_bed文件中有一个区间1 16366079 16509408 1:16366079|16509408 exon,那么如何将1:16366079-16509408内所有外显子的序列提取出来,然后进行拼接。先利用bedtools intersect对exon_bed和gtf文件求一个交集(注意:exon_bed和gtf文件中获取的所有exon区间最好先进行排序),以获得该区间内的所有外显子,如下所示:
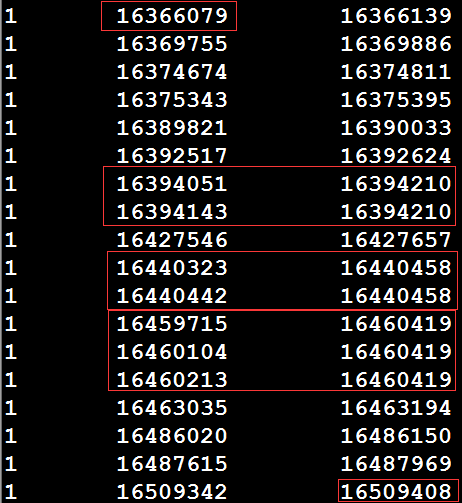
从上图可以看出,由于可变剪切的存在,导致存在多个转录本,从而exon区间存在over_lap的情况,上图需要红框内的行需要进行预处理,选择最大区间。得到下图序列:
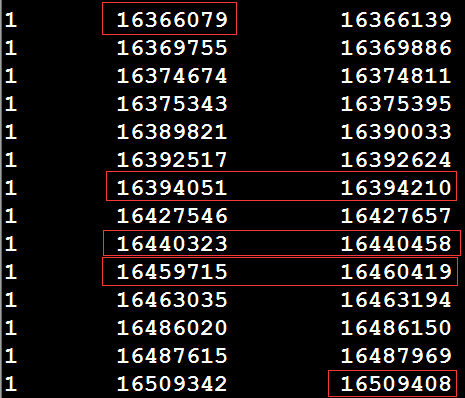
获取最大区间,即bed文件进行处理后,再利用bedtools getfasta从参考序列中提取该区间内的序列,然后进行合并,产生1:16366079-16509408之间所有外显子序列。其中脚本处理如下,使用方法:bash process.sh path_of_refgenome
#!/bin/bash
genome_path=$1
bed_over_path='.' #从Mus_musculus.GRCm38.84.gtf文件中获取所有exon区间,并排序去重
grep -v '^#' $genome_path/Mus_musculus.GRCm38.84.gtf | grep -w 'exon' | cut -f1,4-5 | sort -V | uniq > all_exon_from_gtf if [ ! -d temp ]
then
mkdir temp
fi >temp/exon_all.fa
>exon_all.fa #step1:读取exon_bed的每一行,每一行相当于一个circRNA,将每一行值付给相应变量
#step2:生成过程文件:chrom_start_end_bed记录了exon_bed的每一行信息
#step3:bedtools intersect获取all_exon_from_gtf文件和chrom_start_end_bed交集
#step4:bed_over.py对step3产生的交集chrom_start_end_over_lap.bed文件中的overlap行选取最大外显子区间
#step5:使用bedtools getfasta获取chrom_start_end_over_lap_bed文件对应的fa序列,即完成对exon_bed文件一行的序列提取工作
#step6:进行合并序列操作,先将exon_bed中的>header写入chrom_start_end.fa
#step7:将bedtools getfasta获取的exon序列去掉>行后再去掉换行符,并追加到step6对应的chrom_start_end.fa中
#setp8:最后合并exon_bed文件每一行对应的fa序列,生成exon_all.fa序列文件
while read chrom start end start_end type
do
echo -e "$chrom\t$start\t$end" > "$chrom"_"$start"_"$end"_bed
bedtools intersect -a $genome_path/all_exon_from_gtf -b "$chrom"_"$start"_"$end"_bed > "$chrom"_"$start"_"$end"_over_lap.bed python $bed_over_path/bed_over.py "$chrom"_"$start"_"$end"_over_lap.bed > "$chrom"_"$start"_"$end"_over_lap_bed
bedtools getfasta -fi $genome_path/Mus_musculus.GRCm38.dna.primary_assembly.fa -bed "$chrom"_"$start"_"$end"_over_lap_bed -fo "$chrom"_"$start"_"$end".fa echo ">$start_end" > temp/"$chrom"_"$start"_"$end".fa
grep -v '>' "$chrom"_"$start"_"$end".fa | tr -d "\n" >> temp/"$chrom"_"$start"_"$end".fa
echo '' >> temp/"$chrom"_"$start"_"$end".fa cat temp/"$chrom"_"$start"_"$end".fa >> exon_all.fa
rm -f "$chrom"_"$start"_"$end"_bed
rm -f "$chrom"_"$start"_"$end"_over_lap.bed
rm -f "$chrom"_"$start"_"$end"_over_lap_bed
rm -f "$chrom"_"$start"_"$end".fa
done< exon_bed
其中的bed_over.py脚本是用来处理over_lap行的,脚本见下面:
#!/bin/python
#coding=utf-8
import sys
pre_start = 0
pre_end = 0
flag = 1
pre_chrom = ''
f_open = open(sys.argv[1]) #逐行处理待处理的存在over_lap行的bed文件
#采取的方法是在读取下一行,进行over_lap判断
#需要先保留上一行的chrom,start,end
for each in f_open:
array = each.strip().split('\t')
# chrom = array[3].split(':')[0]
# start = int(array[3].split(':')[1].split('|')[0])
# end = int(array[3].split(':')[1].split('|')[1])
chrom = array[0]
start = array[1]
end = array[2]
if 1 == flag:
pre_start = start
pre_end = end
pre_chrom = chrom
flag = flag + 1
continue
else:
if chrom == pre_chrom:#下一行chrom与上一行相同,才有必要比较是否存在over_lap
if start > pre_end:#下一行start比上一行的end还大,就不存在over_lap,需要将上一行写入文件
#print str(pre_chrom)+'\t'+str(pre_start)+'\t'+str(pre_end)
print '\t'.join([str(pre_chrom),str(pre_start),str(pre_end)])
pre_start = start
pre_end = end
pre_chrom = chrom
flag = flag + 1
continue
elif start <= pre_end:#下一行start比上一行的end小,所以存在over_lap情况,更新end为该行end,替换上一行的end
pre_end = end
pre_chrom = chrom
flag = flag + 1
continue
else:#下一行chrom与上一行chrom不同,直接进行上行写入操作
#print str(pre_chrom)+'\t'+str(pre_start)+'\t'+str(pre_end)
print '\t'.join([str(pre_chrom),str(pre_start),str(pre_end)])
pre_start = start
pre_end = end
pre_chrom = chrom
flag = flag + 1
continue f_open.close()
#最后一行由于没有下一行了,直接进行写入操作
#print str(pre_chrom)+'\t'+str(pre_start)+'\t'+str(pre_end)
print '\t'.join([str(pre_chrom),str(pre_start),str(pre_end)])
circRNA的更多相关文章
- circRNA 中的ALU 重复元件
circRNA 最初研究的很少,只有很小一部分基因有检测到circRNA, 当时都认为是剪切错误形成的,对于其功能也没人去研究:学者对人类的成纤维细胞进行转录组测序,构建去核糖体文库, 同时采用了RN ...
- circRNA 在人和小鼠脑组织中的表达
circRNA 是一类动物体内的内源性的RNA,尽管circRNA的种类丰富,但是其在神经系统中的 功能,并不清楚.科学家通过对人和小鼠的不同脑部组织的RNA 测序,发现了上千种circRNA,经过分 ...
- circRNA研究手册
环状RNA(circRNA)研究技术手册.doc.pdf (转自:汉恒生物)
- 利用circpedia 数据库探究circRNA的可变剪切
circpedia 中收录了利用circexplorer 软件识别到的circRNA, 覆盖了人,小鼠,鸟类,昆虫多个物种的多种细胞系的数据 官网链接如下: http://www.picb.ac.cn ...
- find_circ 识别circRNA 的原理
find_circ 通过识别junction reads 来预测circRNA 和参考基因组比对完之后,首先剔除和基因组完全比对的reads,保留没比对上的reads, 这部分reads 直接比是比对 ...
- CIRI 识别circRNA的原理
CIRI 根据circRNA 连接点处的reads来识别circRNA, 在连接点处的reads 其比对情况非常特殊: CIRI 根据3种模型来识别circRNA, 连接点处的read 叫做junct ...
- circRNA 序列提取中的难点
在预测circRNA时,都是检测breakpoint 处的reads 数,最后给出的环状RNA的ID 都是诸如 chr14:106994222-107183708 这样的形式,给出了起始和终止位置: ...
- CircRNA 环化RNA
2016国自然新秀CircRNA的研究策略和分析
- circRNA数据库的建立
circRNA数据库的建立 wget http://circbase.org/download/human_hg19_circRNAs_putative_spliced_sequence.fa.g ...
随机推荐
- Ajax写成绩批量录入
1.jsp,ajax的循环调用,必须要递归,否则会出错. <%@ page language="java" import="java.util.*" pa ...
- jisuanqi
1.jisuanqi 2.https://github.com/12wangmin/text/tree/master 3.计算截图 7+8 清除 4.总结 通过课程设计,主要要达到两个目的,一是检验和 ...
- 理解java的三大特性之继承
学习来源:http://www.cnblogs.com/chenssy/p/3354884.html default 默认权限(包权限-同一个包可以访问) private 私有(类内部可以使用,继承的 ...
- git学习笔记2——ProGit2
先附上教程--<ProGit 2> 配置信息 Git 自带一个 git config 的工具来帮助设置控制 Git 外观和行为的配置变量. 这些变量存储在三个不同的位置: /etc/git ...
- mysql 8.0,运行springboot项目配置:
1.修改pom.xml <dependency> <groupId>mysql</groupId> <artifactId>mysql-connecto ...
- 洛谷P2698 [USACO12MAR]花盆Flowerpot
P2698 [USACO12MAR]花盆Flowerpot 题目描述 Farmer John has been having trouble making his plants grow, and n ...
- BZOJ4828 AHOI/HNOI2017大佬(动态规划+bfs)
注意到怼大佬的操作至多只能进行两次.我们逐步简化问题. 首先令f[i][j]表示第i天结束后自信值为j时至多有多少天可以进行非防御操作(即恢复自信值之外的操作).这个dp非常显然.由于最终只需要保证存 ...
- MT【43】抛物线不常见性质2.
注:S为抛物线焦点
- BZOJ 4671 异或图 | 线性基 容斥 DFS
题面 Description 定义两个结点数相同的图 G1 与图 G2 的异或为一个新的图 G, 其中如果 (u, v) 在 G1 与 G2 中的出现次数之和为 1, 那么边 (u, v) 在 G 中 ...
- Helm使用详解
使用1.helm search 查看charts stable是官方的 local是自己的 2.查看repo helm repo list 3.安装 helm install stable/mysql ...