【转录组入门】6:reads计数
作业要求:
实现这个功能的软件也很多,还是烦请大家先自己搜索几个教程,入门请统一用htseq-count,对每个样本都会输出一个表达量文件。
需要用脚本合并所有的样本为表达矩阵。参考:生信编程直播第四题:多个同样的行列式文件合并起来
对这个表达矩阵可以自己简单在excel或者R里面摸索,求平均值,方差。
看看一些生物学意义特殊的基因表现如何,比如GAPDH,β-ACTIN等等。
【1】安装计数软件:htseq-count
# conda安装
$ conda install -c bioconda htseq # 测试
# 能够在Python中导入HTSeq这个包,说明安装成功
$ python
>>> import HTSeq
>>>
【2】用htseq对排序后的bam文件进行计数
# 用htseq对排序后的bam文件进行计数
# htseq-count [options] <alignment_file> <gtf_file>
for i in `seq `
do
htseq-count -s no -r pos -f bam RNA-Seq/aligned/SRR35899${i}_sorted.bam reference/gencode.v26lift37.annotation.sorted.gtf > RNA-Seq/matrix/SRR35899${i}.count > RNA-Seq/matrix/SRR35899${i}.log
done # (实际情况:没能对gtf文件进行排序,所以使用未排序的bam文件与原始gtf文件)
for i in `seq `
do
htseq-count -s no -r pos -f bam hisat2_result/SRR35899${i}.bam reference/genome/gencode/gencode.v26lift37.annotation.gtf > htseq-count_result/SRR35899${i}.count >log_SRR35899${i}_htseq
done
-f bam/sam: 指定输入文件格式,默认SAM
-r name/pos: 你需要利用samtool sort对数据根据read name或者位置进行排序,默认是name
-s yes/no/reverse: 数据是否来自于strand-specific assay。DNA是双链的,所以需要判断到底来自于哪条链。如果选择了no, 那么每一条read都会跟正义链和反义链进行比较。默认的yes对于双端测序表示第一个read都在同一个链上,第二个read则在另一条链上。
程序运行结束后得到矩阵文件:XXXXX.count
【3】合并表达矩阵
# R脚本
# . 导入数据
# 首先将四个文件分别赋值:control1,control2,rep1,rep2
>control1 <- read.table("~/disk2/data/rna-seq/matrix/SRR3589959.count", sep="\t", col.names = c("gene_id","control1"))
>control2 <- read.table("~/disk2/data/rna-seq/matrix/SRR3589961.count", sep="\t", col.names = c("gene_id","control2"))
>rep1 <- read.table("~/disk2/data/rna-seq/matrix/SRR3589960.count", sep="\t", col.names = c("gene_id","treat1"))
>rep2 <- read.table("~/disk2/data/rna-seq/matrix/SRR3589962.count", sep="\t",col.names = c("gene_id","treat2"))
# . 数据整合
# 将四个矩阵按照gene_id进行合并,并赋值给raw_count
>raw_count <- merge(merge(control1, contol2, by="gene_id"), merge(rep1,rep2, by="gene_id"))
# 去掉前五行
>raw_count_filter <- raw_count[-:-, ]
# 因为我们无法在EBI数据库上直接搜索找到ENSMUSG00000024045.5这样的基因,只能是ENSMUSG00000024045的整数,没有小数点,所以需要进一步替换为整数的形式。
# 第一步将匹配到的.以及后面的数字连续匹配并替换为空,并赋值给ENSEMBL
>ENSEMBL <- gsub("\\.\\d*", "", raw_count_filter$gene_id)
# 将ENSEMBL重新添加到raw_count_filt1矩阵
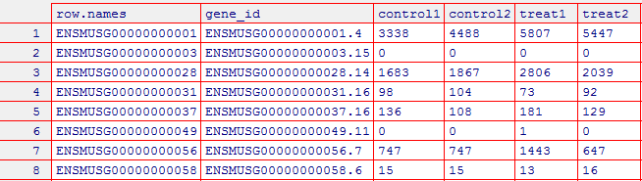
> row.names(raw_count_filter) <- ENSEMBL
# . 查看数据整体情况
> summary(raw_count_filter)
# . 将raw_count_filter写入csv文件(可用Excel文件)
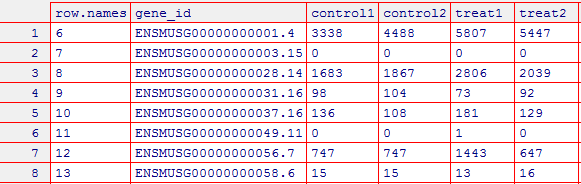
> write.csv(raw_count_filter,"d:/cs_file/r_file/file-lyx0623/H_N_rawcount.csv",row.names=FALSE)

理论知识
关于比对:
】想知道已知基因的表达情况:选择alignment-free工具(例如salmon, sailfish)
】想找noval isoforms:alignment-based工具(如HISAT2, STAR)
】转录本定量:需要考虑更多的因素
1、基因表达定量有3个水平:基因水平、转录本水平、外显子使用水平

基因水平:
Htseq-count等工具作用:根据read和基因位置的overlap,判断该read属于哪个基因
转录本水平:
Cufflinks等工具处理的难题:转录本亚型(isoforms)之间通常是有重叠的,当二代测序读长低于转录本长度时,如何进行区分?这些工具大多采用的都是expectation maximization(EM)。好在我们有三代测序。
外显子使用水平:与基因水平类似。为了更好的计数,需要提供无重叠的外显子区域的gtf文件,用于分析差异外显子使用的DEXSeq提供了一个Python脚本(dexseq_prepare_annotation.py)执行这个任务。
2、reads计数后得到的基因定量结果(count matrix),在进行不同维度的比较时需要进行不同的处理(标准化):
【】比较同一个样本(within-sample)不同基因之间的表达情况---主要考虑转录本长度的影响
(因为转录本越长,那么检测的片段也会更多,直接比较等于让小孩和大人进行赛跑)
【】比较不同样本(across-sample)同一个基因的表达情况---主要考虑测序深度的影响
(测序深度越高,检测到的概率越大)
标准化的算法:RPKM(SE), FPKM(PE),TPM, TMM,RSEM等等。
一般,标准化之后才能进行差异表达分析(某些软件要求原始数据,未经标准化)
htseq参数说明
用法:htseq-count [options] alignment_file gff_file
alignment_file:比对结果文件,可以是sam格式或bam格式,可以通过后面介绍的-f参数指定,默认是sam格式。推荐使用按照name排序后的sam文件,这样的好处是消耗的内存更少,效率更高。关于输入文件排序详见参数-r。
gff_file: 包含单位信息的gff/GTF文件(gff文件格式),大多数情况下就是指注释文件; 由于GTF文件其实就是gff文件格式的变形,在这里同样可以传入GTF格式文件。
-f bam/sam: 指定输入文件格式,默认SAM
-r name/pos: 你需要利用samtool sort对数据根据read name或者位置进行排序,默认是name
-s yes/no/reverse: 数据是否来自于strand-specific assay。DNA是双链的,所以需要判断到底来自于哪条链。如果选择了no, 那么每一条read都会跟正义链和反义链进行比较。默认的yes对于双端测序表示第一个read都在同一个链上,第二个read则在另一条链上。
-a 最低质量, 剔除低于阈值的read
-m 模式 union(默认), intersection-strict and intersection-nonempty。一般而言就用默认的,作者也是这样认为的。
-i id attribute: 在GTF文件的最后一栏里,会有这个基因的多个命名方式(如下), RNA-Seq数据分析常用的是gene_id, 当然你可以写一个脚本替换成其他命名方式。
【转录组入门】6:reads计数的更多相关文章
- 【转录组入门】3:了解fastq测序数据
操作:需要用安装好的sratoolkit把sra文件转换为fastq格式的测序文件,并且用fastqc软件测试测序文件的质量 作业:理解测序reads,GC含量,质量值,接头,index,fastqc ...
- Storm入门2-单词计数案例学习
[本篇文章主要是通过一个单词计数的案例学习,来加深对storm的基本概念的理解以及基本的开发流程和如何提交并运行一个拓扑] 单词计数拓扑WordCountTopology实现的基本功能就是不停地读入 ...
- 弗雷塞斯 从生物学到生物信息学到机器学习 转录组入门(3):了解fastq测序数据
sra文件转换为fastq格式 1 fastq-dump -h --split-3 也就是说如果SRA文件中只有一个文件,那么这个参数就会被忽略.如果原文件中有两个文件,那么它就会把成对的文件按*_1 ...
- 转录组入门(3):了解fastq测序数据
sra文件转换为fastq格式 fastq-dump -h --split-3 也就是说如果SRA文件中只有一个文件,那么这个参数就会被忽略.如果原文件中有两个文件,那么它就会把成对的文件按*_1.f ...
- Bulk RNA-Seq转录组学习
与之对应的是single cell RNA-Seq,后面也会有类似文章. 参考:https://github.com/xuzhougeng/Learn-Bioinformatics/ 作业:RNA-s ...
- RNA-seq数据综合分析教程 AKAP95
https://blog.csdn.net/l_yivs?t=1 RNA-seq数据综合分析教程 2 4,055 A+ 所属分类:Transcriptomics 收 藏 2 RNA-se ...
- 【转载】Spark性能优化指南——高级篇
前言 数据倾斜调优 调优概述 数据倾斜发生时的现象 数据倾斜发生的原理 如何定位导致数据倾斜的代码 查看导致数据倾斜的key的数据分布情况 数据倾斜的解决方案 解决方案一:使用Hive ETL预处理数 ...
- 【转】【技术博客】Spark性能优化指南——高级篇
http://mp.weixin.qq.com/s?__biz=MjM5NjQ5MTI5OA==&mid=2651745207&idx=1&sn=3d70d59cede236e ...
- NOIP2017 国庆郑州集训知识梳理汇总
第一天 基础算法&&数学 day1难度测试 如果要用一个词来形容上午的测试,那真是体无完肤. 成绩: 题目 成绩 评价 T1 50 一般 T2 10 大失所望 T3 0 差 基础算法 ...
随机推荐
- AVL Tree Deletion
Overview 知识点: 1. delete函数的signature public AVLTreeNode Delete(AVLTreeNode node, int key) 2. 算法,如何删除节 ...
- jQuery-4.动画篇---淡入淡出效果
jQuery中淡出动画fadeOut 让元素在页面不可见,常用的办法就是通过设置样式的display:none.除此之外还可以一些类似的办法可以达到这个目的.这里要提一个透明度的方法,设置元素透明度为 ...
- Linux 常用命令笔记-2
注意事项: 沟通项目需求:1.项目背景和目的 哪个团队.项目Wiki? 数据库登陆:mysql -uroot -pabc@0326 -h127.0.0.1 -P4004 -A set names ut ...
- VIM:Found a swap file by the name
在linux下用vi或vim打开Test.java文件时 [root@localhost tmp]# vi Test.java出现了如下信息: E325: ATTENTION Found a s ...
- Elasticsearch CURL命令
1.查看集群状态 curl '10.18.37.223:9200/_cat/health?v'绿色表示一切正常, 黄色表示所有的数据可用但是部分副本还没有分配,红色表示部分数据因为某些原因不可用 2. ...
- AI之旅(1):出发前的热身运动
前置知识 无 知识地图 自学就像在海中游泳 当初为什么会想要了解机器学习呢,应该只是纯粹的好奇心吧.AI似乎无处不在,又无迹可循.为什么一个程序能在围棋的领域战胜人类,程序真的有那么聪明吗?如 ...
- Python3.7 练习题(三) 将指定目录下的图片进行批量尺寸大小处理
# 将指定目录下的图片进行批量尺寸大小处理 #修改图片尺寸 导入Image os 快捷键 alt+enter import os from PIL import Image def process_i ...
- 关于OSI
OSI模型,即开放式通信系统互联参考模型(Open System Interconnection,OSI/RM,Open Systems Interconnection Reference Model ...
- 《DSP using MATLAB》Problem 7.26
注意:高通的线性相位FIR滤波器,不能是第2类,所以其长度必须为奇数.这里取M=31,过渡带里采样值抄书上的. 代码: %% +++++++++++++++++++++++++++++++++++++ ...
- django from表单验证
django from表单验证 实现:表单验证 工程示例: urls.py 1 2 3 4 5 6 7 8 9 from django.conf.urls import url from djan ...