hisat2+stringtie+ballgown
hisat2+stringtie+ballgown
早在去年九月,我就写个博文说 RNA-seq流程需要进化啦!http://www.bio-info-trainee.com/1022.html ,主要就是进化成hisat2+stringtie+ballgown的流程,但是我一直没有系统性的讲这个流程,因为我觉真心木有用。我只用了里面的hisat来做比对而已!但是群里的小伙伴问得特别多,我还是勉为其难的写一个教程吧,你们之间拷贝我的代码就可以安装这些软件的!然后自己找一个测试数据,我的脚本很容易用的!
## Download and install HISAT# https://ccb.jhu.edu/software/hisat2/index.shtmlcd ~/biosoftmkdir HISAT && cd HISAT#### readme: https://ccb.jhu.edu/software/hisat2/manual.shtmlwget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/downloads/hisat2-2.0.4-Linux_x86_64.zipunzip hisat2-2.0.4-Linux_x86_64.zipln -s hisat2-2.0.4 current## ~/biosoft/HISAT/current/hisat2-build## ~/biosoft/HISAT/current/hisat2## Download and install StringTie## https://ccb.jhu.edu/software/stringtie/ ## https://ccb.jhu.edu/software/stringtie/index.shtml?t=manualcd ~/biosoftmkdir StringTie && cd StringTiewget http://ccb.jhu.edu/software/stringtie/dl/stringtie-1.2.3.Linux_x86_64.tar.gztar zxvf stringtie-1.2.3.Linux_x86_64.tar.gzln -s stringtie-1.2.3.Linux_x86_64 current# ~/biosoft/StringTie/current/stringtie
while read iddosample=$(echo $id |cut -d" " -f 1 )file1=$(echo $id |cut -d" " -f 2 )file2=$(echo $id |cut -d" " -f 3 )echo $sampleecho $file1echo $file2~/biosoft/HISAT/current/hisat2 -p 4 --dta -x ~/reference/index/hisat/hg19/genome -1 $file1 -2 $file2 -S $sample.hisat2.hg19.sam 2>$sample.hisat2.hg19.log &done <$1

while read iddofile=$(basename $id )sample=${file%%.*}echo $id $samplenohup samtools sort -@ 4 -o ${sample}.sorted.bam $id &done <$1

while read iddofile=$(basename $id )sample=${file%%.*}echo $id $samplenohup ~/biosoft/StringTie/current/stringtie -p 4 -G ~/reference/gtf/gencode/gencode.v25lift37.annotation.gtf -o $sample.hg19.stringtie.gtf -l $sample $id &done <$1

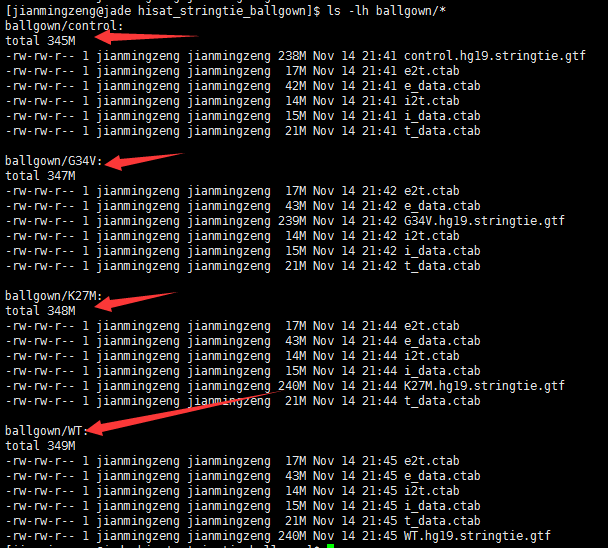
This entry was posted in 转录组软件 and tagged ballgown, hisat2, StringTie, 转录组 by ulwvfje. Bookmark the permalink.
hisat2+stringtie+ballgown的更多相关文章
- 转录组分析---Hisat2+StringTie+Ballgown使用
转录组分析---Hisat2+StringTie+Ballgown使用 (2016-10-10 08:14:45) 转载▼ 标签: 生物信息学 转录组 1.Hisat2建立基因组索引: First ...
- HISAT2+StringTie+Ballgown安装及使用流程
HISAT2+StringTie+Ballgown安装及使用流程 2015年Nature Methods上面发表了一款快速比对工具hisat,作为接替tophat和bowtie的比对工具,它具有更快的 ...
- HISAT2,StringTie,Ballgown处理转录组数据
HISAT2,StringTie,Ballgown处理转录组数据 本文总阅读量次2017-05-26 HISAT2,StringTie,Ballgown处理转录组数据思路如下: 数据质控 将RNA-s ...
- NGS NGS ngs(hisat,stringtie,ballgown)
NGS ngs(hisat,stringtie,ballgown) #HISAT (hierarchical indexing for spliced alignment of transcripts ...
- HISAT,sTRINGTIE,ballgown三款RNA-seq信息分析软件
HISAT,sTRINGTIE,ballgown三款RNA-seq信息分析软件 2015年04月02日 11:35:47 夜丘 阅读数:8940 标签: 生物 更多 个人分类: 论文笔记 Bowt ...
- 转录组的组装Stingtie和Cufflinks
转录组的组装Stingtie和Cufflinks Posted: 十月 18, 2017 Under: Transcriptomics By Kai no Comments 首先这两款软件都是用 ...
- StringTie用法详解
StringTie 参考链接: https://ccb.jhu.edu/software/stringtie/index.shtml?t=manual#input https://www.cnblog ...
- 转录本组装软件StringTie的使用说明
转录本组装软件StringTie的使用说明 StringTie 转录本组装软件StringTie的使用说明 转录组分析流程 HISTA + StringTie 组合.其Protocol 发表在Natu ...
- 转录组差异表达分析工具Ballgown
Ballgown是分析转录组差异表达的R包. 软件安装: 运行R, source(“http://bioconductor.org/biocLite.R”) biocLite(“ballgown”) ...
随机推荐
- vue如果是首页了 不让其后退
history.pushState(null, null, document.URL); //首页加载时候先置空 window.addEventListener('popstate', functio ...
- ftp 传输问题
服务器配置ftp站点后,客户端机器可以下载但不能上传文件? 今天从公网的服务器连接本地内网的FTP server copy文件时,系统老是提示227 Entering Passive Mode (xx ...
- jquery val() text() html()的区别
value()主要用在表单元素上,如果其他的元素获取value是通过attract()的方法,text()是获取元素的纯文本,如果text(“content”)就会更改元素的文本内容:html()获取 ...
- springMVC :interceptors
1.配置拦截器 在springMVC.xml配置文件增加: <mvc:interceptors> <!-- 日志拦截器 --> <mvc:interc ...
- Feign 注意事项
一.FeignClient注解 FeignClient注解被@Target(ElementType.TYPE)修饰,表示FeignClient注解的作用目标在接口上 1 2 3 4 5 @FeignC ...
- 游戏AI技术 2
[Unity3D人工智能编程精粹 2] 1.跟随领队行为. 用靠近(Seek)或追逐(Pursuit)实现跟随领队行为并不好.在Seek中,AI角色会被推向领队,最终与领队占据相同位置.而Pursui ...
- Avatar
[Avatar] 1.Retargeting of Humanoid animations Retargeting is only possible for humanoid models, wher ...
- NBU 还原LINUX ORACLE 数据库(EHR)
一.E-HR数据库(全备)恢复 目录 一.E-HR数据库(全备)恢复... 1 1. 使用bplist 命令读取备份文件... 1 2. 启动到nomount状态... 2 3. 利用rman还原控制 ...
- as3.0 在数组中找个找个,并且替换
var arr:Array=[1,2,7,9,3,5,6]; var findNum:Number =5//想要找到的数字 var replaceNum:Object =3//想要替换的数字 var ...
- Gym - 100989G 二分
链接:ECJTU 2018 Summer Training 1 - Virtual Judge https://vjudge.net/contest/236677#problem/G 谷歌翻译: 距 ...