QIIME2使用方法
激活qiime2的执行环境:source activate qiime2-2019.4
如何查看conda已有的环境:conda info -e 以下分析流程参考:https://docs.qiime2.org/2019.4/tutorials/qiime2-for-experienced-microbiome-researchers/
1、数据准备
现在我们常用的就是这种格式的数据,每个样品一对数据文件
数据来源:https://docs.qiime2.org/2019.7/tutorials/importing/
wget \
-O "casava-18-paired-end-demultiplexed.zip" \
"https://data.qiime2.org/2019.4/tutorials/importing/casava-18-paired-end-demultiplexed.zip" 下载解压后,文件夹中文件如下:
2、将数据转换为qza格式(qiime新定义的自己的格式类型,有点编程中对象的含义)
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \
--input-path casava-18-paired-end-demultiplexed \
--input-format CasavaOneEightSingleLanePerSampleDirFmt \
--output-path demux-paired-end.qza 3、查看数据质量
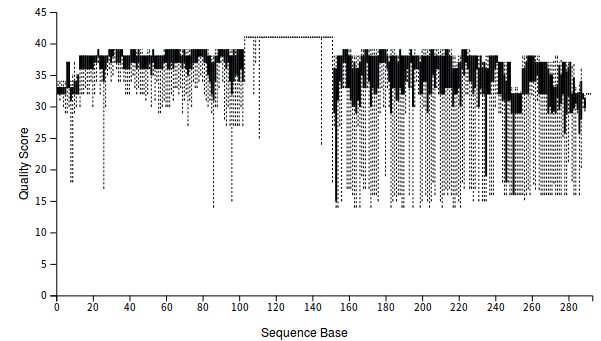
qiime demux summarize --i-data demux-paired-end.qza --o-visualization demux-summary-1.qzv
用以下命令查看结果:
qiime tools view demux-summary-1.qzv
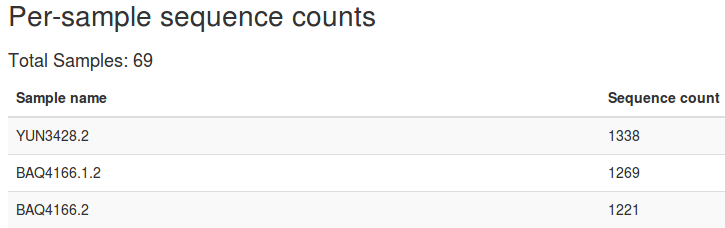

4、双端序列合并成单端
qiime vsearch join-pairs --i-demultiplexed-seqs demux-paired-end.qza --o-joined-sequences demux-joinded.qza

5、查看对merge后的数据质量情况
qiime demux summarize --i-data demux-joinded.qza --o-visualization demux-summary-merged.qzv
qiime tools view demux-summary-merged.qzv
##### 以下是使用dada2进行数据去噪,本教程先跳过该步,之后有专门教程介绍dada2使用
4、对数据进行剪切
双端:
qiime dada2 denoise-paired \
--i-demultiplexed-seqs demux-paired-end.qza \
--p-trim-left-f 13 \
--p-trim-left-r 13 \
--p-trunc-len-f 150 \
--p-trunc-len-r 150 \
--o-table table.qza \
--o-representative-sequences rep-seqs.qza \
--o-denoising-stats denoising-stats.qza
单端:
qiime dada2 denoise-single \
--i-demultiplexed-seqs demux-joinded.qza \ #输入应该也是序列,不能是joined对象
--p-trim-left 13 \
--p-trunc-len 150 \
--o-table table.qza \
--o-representative-sequences rep-seqs-merged.qza \
--o-denoising-stats denoising-stats-merged.qza
https://forum.qiime2.org/t/demultiplexing-and-trimming-adapters-from-reads-with-q2-cutadapt/2313
以下参考:
https://blog.csdn.net/woodcorpse/article/details/86685524
https://mp.weixin.qq.com/s/6cLzyJjWQmHm82_U6euJ1g
5、序列质控
qiime quality-filter q-score-joined \
--i-demux demux-joinded.qza \
--o-filtered-sequences demux-joined-filtered.qza \
--o-filter-stats demux-joined-filter-stats.qza
输出结果:
- demux-joined-filter-stats.qza: 统计结果
- demux-joined-filtered.qza: 数据过滤后结果
6、用deblur去冗余,并生成特征表(相当于QIIME1的OTU Table)
qiime deblur denoise-16S \
--i-demultiplexed-seqs demux-joined-filtered.qza \
--p-trim-length 250 \
--p-sample-stats \
--o-representative-sequences rep-seqs.qza \
--o-table table.qza \
--o-stats deblur-stats.qza
输出结果:
- rep-seqs.qza: 代表序列
- deblur-stats.qza: 统计过程
- table.qza: 特征表
备注:
由于DADA2和Deblur产生的“OTU”是通过对唯一序列进行分组而创建的,因此这些OTU相当于来自QIIME 1的100%相似度的OTU,通常称为序列变体。在QIIME 2中,这些OTU比QIIME 1默认的97%相似度聚类的OTU具有更高的分辨率,并且它们具有更高的质量,因为这些质量控制步骤比QIIME 1中实现更好。因此,与QIIME 1相比,可以对样本的多样性和分类组成进行更准确的估计。
7、查看deblur去冗余后的特征表
qiime feature-table summarize \
--i-table table.qza \
--o-visualization table.qzv
--m-sample-metadata-file sample-metadata.tsv
qiime feature-table tabulate-seqs \
--i-data rep-seqs.qza \
--o-visualization rep-seqs.qzv
qiime tools view table.qzv
8、统计每个样品包含的序列数
qiime deblur visualize-stats \
--i-deblur-stats deblur-stats.qza \
--o-visualization deblur-stats.qzv
qiime tools view deblur-stats.qzv
9、构建进化树用于多样性分析
qiime phylogeny align-to-tree-mafft-fasttree \
--i-sequences rep-seqs.qza \
--o-alignment aligned-rep-seqs.qza \
--o-masked-alignment masked-aligned-rep-seqs.qza \
--o-tree unrooted-tree.qza \
--o-rooted-tree rooted-tree.qza
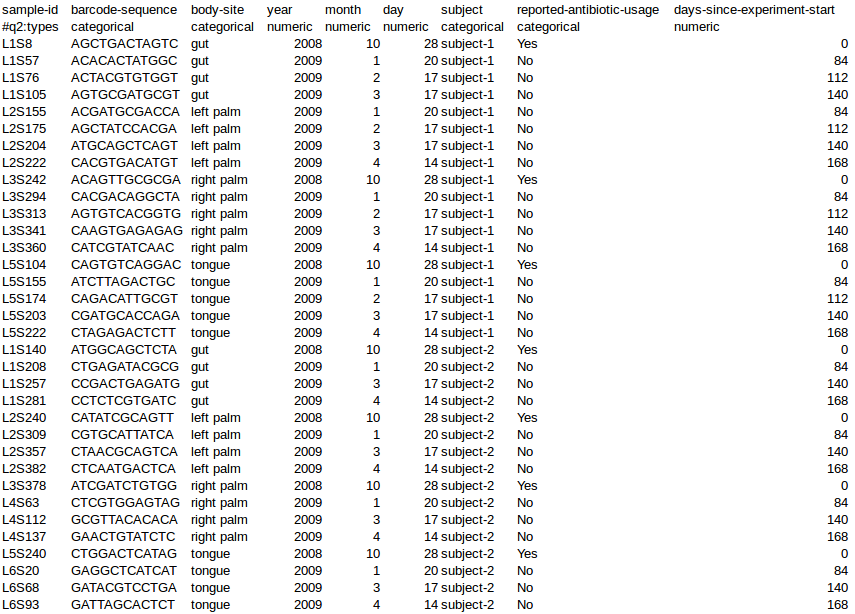
10、需要先准备一个metadata文件,文件说明参考:https://docs.qiime2.org/2019.7/tutorials/metadata/
11、计算核心多样性
qiime diversity core-metrics-phylogenetic \
--i-phylogeny rooted-tree.qza \
--i-table table.qza \
--p-sampling-depth 500 \
--m-metadata-file sample-metadata.tsv \
--output-dir core-metrics-results
分析结果包含:
α多样性
香农(Shannon’s)多样性指数(群落丰富度的定量度量,即包括丰富度
richness和均匀度evenness两个层面)Observed OTUs(群落丰富度的定性度量,只包括丰富度)
Faith’s系统发育多样性(包含特征之间的系统发育关系的群落丰富度的定性度量)
均匀度(或 Pielou’s均匀度;群落均匀度的度量)
β多样性
Jaccard距离(群落差异的定性度量,即只考虑种类,不考虑丰度)
Bray-Curtis距离(群落差异的定量度量)
非加权UniFrac距离(包含特征之间的系统发育关系的群落差异定性度量)
加权UniFrac距离(包含特征之间的系统发育关系的群落差异定量度量)
β多样性分析结果-PCoA:
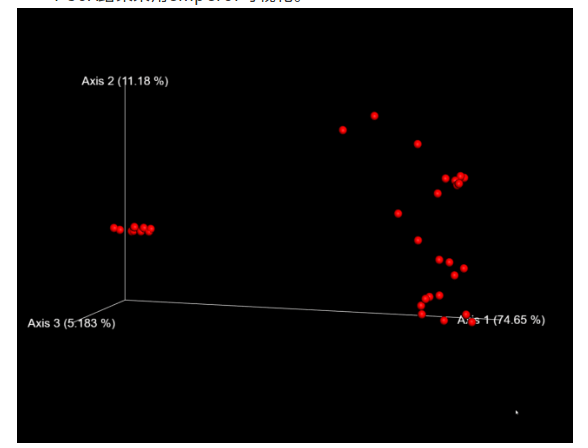
12、Alpha多样性组间显著性分析和可视化
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/faith_pd_vector.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization core-metrics-results/faith-pd-group-significance.qzv
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/evenness_vector.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization core-metrics-results/evenness-group-significance.qzv
13、绘制稀疏曲线
qiime diversity alpha-rarefaction \
--i-table table.qza \
--i-phylogeny rooted-tree.qza \
--p-max-depth 1000 \
--m-metadata-file sample-metadata.tsv \
--o-visualization alpha-rarefaction.qzv
--p-max-depth参数的值应该通过查看上面创建的table.qzv文件中呈现的“每个样本的测序量”信息来确定。一般来说,选择一个在中位数附近的值似乎很好用。
14、物种组成分析
下载物种注释数据库制作的分类器:
wget \
-O "gg-13-8-99-515-806-nb-classifier.qza" \
"https://data.qiime2.org/2018.11/common/gg-13-8-99-515-806-nb-classifier.qza"
物种注释和可视化
qiime feature-classifier classify-sklearn \
--i-classifier gg-13-8-99-515-806-nb-classifier.qza \
--i-reads rep-seqs.qza \
--o-classification taxonomy.qza
qiime metadata tabulate \
--m-input-file taxonomy.qza \
--o-visualization taxonomy.qzv
生成物种组成柱状图:
qiime taxa barplot \
--i-table table.qza \
--i-taxonomy taxonomy.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization taxa-bar-plots.qzv

QIIME2使用方法的更多相关文章
- javaSE27天复习总结
JAVA学习总结 2 第一天 2 1:计算机概述(了解) 2 (1)计算机 2 (2)计算机硬件 2 (3)计算机软件 2 (4)软件开发(理解) 2 (5) ...
- 扩增子分析QIIME2. 1简介和安装
原网站:https://blog.csdn.net/woodcorpse/article/details/75103929 声明:本文为QIIME2官方帮助文档的中文版,由中科院遗传发育所刘永鑫博士翻 ...
- mapreduce多文件输出的两方法
mapreduce多文件输出的两方法 package duogemap; import java.io.IOException; import org.apache.hadoop.conf ...
- 【.net 深呼吸】细说CodeDom(6):方法参数
本文老周就给大伙伴们介绍一下方法参数代码的生成. 在开始之前,先补充一下上一篇烂文的内容.在上一篇文章中,老周检讨了 MemberAttributes 枚举的用法,老周此前误以为该枚举不能进行按位操作 ...
- IE6、7下html标签间存在空白符,导致渲染后占用多余空白位置的原因及解决方法
直接上图:原因:该div包含的内容是靠后台进行print操作,输出的.如果没有输出任何内容,浏览器会默认给该空白区域添加空白符.在IE6.7下,浏览器解析渲染时,会认为空白符也是占位置的,默认其具有字 ...
- 多线程爬坑之路-Thread和Runable源码解析之基本方法的运用实例
前面的文章:多线程爬坑之路-学习多线程需要来了解哪些东西?(concurrent并发包的数据结构和线程池,Locks锁,Atomic原子类) 多线程爬坑之路-Thread和Runable源码解析 前面 ...
- [C#] C# 基础回顾 - 匿名方法
C# 基础回顾 - 匿名方法 目录 简介 匿名方法的参数使用范围 委托示例 简介 在 C# 2.0 之前的版本中,我们创建委托的唯一形式 -- 命名方法. 而 C# 2.0 -- 引进了匿名方法,在 ...
- ArcGIS 10.0紧凑型切片读写方法
首先介绍一下ArcGIS10.0的缓存机制: 切片方案 切片方案包括缓存的比例级别.切片尺寸和切片原点.这些属性定义缓存边界的存在位置,在某些客户端中叠加缓存时匹配这些属性十分重要.图像格式和抗锯齿等 ...
- [BOT] 一种android中实现“圆角矩形”的方法
内容简介 文章介绍ImageView(方法也可以应用到其它View)圆角矩形(包括圆形)的一种实现方式,四个角可以分别指定为圆角.思路是利用"Xfermode + Path"来进行 ...
随机推荐
- lua字符串分割函数[适配中文特殊符号混合]
lua的官方函数里无字符串分割,起初写了个简单的,随之发现如果是中文.字符串.特殊符号就会出现分割错误的情况,所以就有了这个zsplit. function zsplit(strn, chars) f ...
- Ubuntu 18.04 64位安装tensorflow-gpu
第一步(可直接跳到第二步):安装nvidia显卡驱动 linux用户可以通过官方ppa解决安装GPU驱动的问题.使用如下命令添加Graphic Drivers PPA: 1 sudo add-apt- ...
- 【题解】BZOJ4883: [Lydsy1705月赛]棋盘上的守卫(最小生成基环森林)
[题解]BZOJ4883: [Lydsy1705月赛]棋盘上的守卫(最小生成基环森林) 神题 我的想法是,每行每列都要有匹配且一个点只能匹配一个,于是就把格点和每行每列建点出来做一个最小生成树,但是不 ...
- JVM之GC回收信息详解
一.-XX:+PrintGCDetails 打印GC日志 参数配置:-Xms10M -Xmx10M -XX:+PrintGCDetails -XX:+PrintGCDateStamps -XX:+Pr ...
- Head First设计模式——组合模式
最近比较忙,有段时间没有更新设计模式的进度了.今天继续学习组合设计模式. 组合模式的例子我们继续延续上篇<Head First设计模式——迭代器模式>的菜单例子,首先声明下迭代器和组合模式 ...
- 用python做推荐系统(一)
一.简介: 推荐系统是最常见的数据分析应用之一,包含淘宝.豆瓣.今日头条都是利用推荐系统来推荐用户内容.推荐算法的方式分为两种,一种是根据用户推荐,一种是根据商品推荐,根据用户推荐主要是找出和这个用户 ...
- SpringBoot中SpringMVC的自动配置以及扩展
一.问题引入 我们在SSM中使用SpringMVC的时候,需要由我们自己写SpringMVC的配置文件,需要用到什么就要自己配什么,配置起来也特别的麻烦.我们使用SpringBoot的时候没有进行配置 ...
- OSS上传图片无法在线预览的解决方案
OSS上传图片无法在线预览的解决方案 最近在做的项目涉及到商品详情,由于前端用的flutter框架并且该详情为富文本,dart语言关于富文本的组件不是非常友好,当富文本中的图片无法在浏览器中直接预览的 ...
- Cassandra数据建模中最重要的事情:主键
Cassandra数据建模中要了解的最重要的事情:主键 使用关系数据建模,您可以从主键开始,但是RDBMS中的有效数据模型更多地是关于表之间的外键关系和关系约束.由于Cassandra无法使用JOIN ...
- String类方法的使用
String类的判断功能: boolean equals(Object obj) //比较字符串内容是否相同(区分大小写). boolean equalsIgnoreCase(String str) ...