Journal of Proteome Research | Clinically Applicable Deep Learning Algorithm Using Quantitative Proteomic Data (分享人:翁海玉)
题目:Clinically Applicable Deep Learning Algorithm Using Quantitative Proteomic Data
期刊:Journal of Proteome Research
发表时间:August 2, 2019
DOI:: 10.1021/acs.jproteome.9b00268
分享人:翁海玉
内容与观点:
本研究描述了一种优化的基于深度学习(DL)的胰腺癌诊断方法并测试了该方法的分类能力。
1、实验设计
1.1数据集构建:该方法使用1008个样本的选择反应监测-质谱(SRM - MS)数据集,SRM-MS在血浆样本中检测出34种多肽(由34个蛋白衍生而来)。数据集包括300个正常人样本(NC),109个胰腺癌良性样本(PB),49个其他良性样本(OB),149个其他癌症样本(OC),和401个胰腺癌样本(PDAC)。按照0.7:0.3的比例将数据集分为训练集(691 samples; 322 PDAC, 41 OB, 88 PB, and 240 NC)和测试集(317 samples; 79 PDAC, 8 OB, 149 OC, 21 PB, and 60 NC),保持内部比例不变。其中OC只在测试集中有,以确定是否构建的模型会受到癌症异质性影响。
为了算法能够表现出鉴别胰腺癌的能力,数据集被重新构建为控制组(NC+PB+OB+OC),病例组(PDAC)。
1.2 DL模型训练和参数优化:采用十倍交叉验证的方法对训练数据集进行处理,避免了抽样偏差。每次迭代从子训练数据集中随机抽取约622个数据点(691*0.9)输入模型;其余69个值(691*0.1)作为子测试数据集,用于评估模型中的误差,同时对每个选定的数据点(分层抽样)保持对照组和病例组的比例相等。为了构造该模型,我们采用逐步逼近的方法来减少测试所有可能特征集的计算量。
利用训练数据集对模型进行微调,优化参数。然后在独立的测试数据集上对训练后的模型进行测试,并对其分类性能进行评估。利用独立的测试数据集进一步验证了模型的性能。利用测试数据集的性能来指导参数的优化。为了减少样本选择偏差和模型过拟合的可能性,除了交叉验证外,还进行了bootstrapping验证。
训练和测试数据集使用v3.10.3.6版本的H2O软件包进行处理。DL方法对10个最重要的参数纪元数(number of epochs)、节点数和隐层数(number of nodes and hidden layers)、激活函数(activation function)、rho、epsilon、L1 & L2正则化(L1 & L2 regularization)、隐藏丢失率(hidden dropout ratio)、输入丢失率(input dropout ratio)、每次迭代训练样本(train samples per iteration)、最大w2(max w2)。同时进行网格搜索来优化每个参数的值。并使用每个参数的常用值对它们逐一进行了优化,以此确定重要参数。
1.3 五种传统机器学习模型参数优化:对在蛋白质组学应用最广泛的五种机器学习模型:随机森林(RF)、支持向量机(SVM),逻辑回归(LR),K近邻(KNN)和贝叶斯(NB)建模,训练和测试数据集的处理与DL方法相同。用网格搜索,对5种方法中的参数进行调优。
1.4 DL与传统模型比较:
采用了五种传统的模型性能指标:查全率、精密度、F1评分、精密度和工作特性曲线下面积(AUROC):
Recall= 
Precision= 
F1 score= 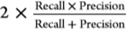
Accuracy= 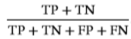
AUROC通过测量这个图的recall和FDR来构建AUROC曲线,其中1.0表示完全分离,0.5表示随机分类。如图:
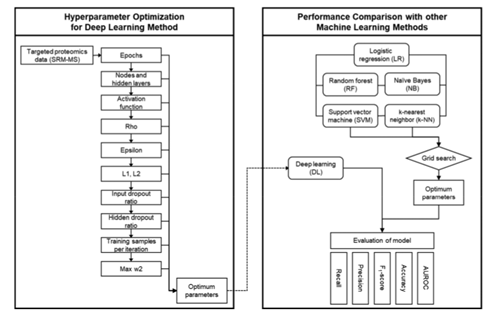
Figure 1 完整实验过程
2、结果
2.1 DL参数优化:10个参数中 epoch, activation function, epsilon, input dropout ratio影响DL模型的分类模型(Figure 2 ),如图,选择了AUROC最大时的值为参数值。
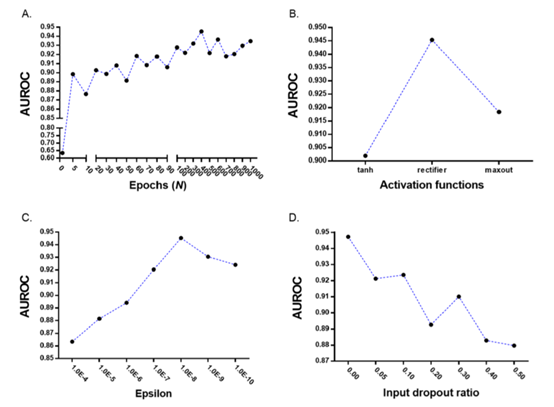
Figure 2 DL参数优化
2.2 DL与传统机器学习模型比较:
各个指标都有明显提升,如下图:
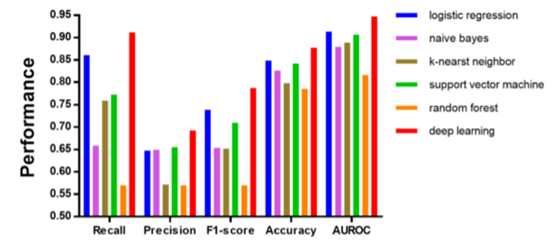
Figure 3 六个模型的性能参数柱状图
3、结论
研究结果表明,DL是蛋白组学数据生物标志物确认的有力工具。在临床实验室中,DL有提高疾病分类任务的标准化和内部可靠性的潜力。未来的工作应该优化其在临床环境中的表现,以充分利用DL方法作为临床工具。
4、讨论
虽然DL各个性能指标都远远高于传统方法,但其仍然存在耗时长,电脑硬件要求高,需要更多的特征和样本的数据集等局限,尤其受到质疑的是,DL是一个黑盒子,难以给出内部过程。但本文向我们展示了DL的潜力。相信DL预测不同群体的高精度的能力将产生全新的数据处理选项,支持和加强未来基于蛋白组学的生物标志物研究。
Journal of Proteome Research | Clinically Applicable Deep Learning Algorithm Using Quantitative Proteomic Data (分享人:翁海玉)的更多相关文章
- Journal of Proteome Research | Improving Silkworm Genome Annotation Using a Proteogenomics Approach (分享人:张霞)
题目:Improving Silkworm Genome Annotation Using a Proteogenomics Approach 期刊:Journal of Proteome Resea ...
- Integrative Analysis of MicroRNAome, Transcriptome, and Proteome during the Limb Regeneration of Cynops orientalis (文献分享一组-翁海玉)
文献名:Integrative Analysis of MicroRNAome, Transcriptome, and Proteome during the Limb Regeneration of ...
- Journal of Proteome Research | 人类牙槽骨蛋白的蛋白质组学和n端分析:改进的蛋白质提取方法和LysargiNase消化策略增加了蛋白质组的覆盖率和缺失蛋白的识别 | (解读人:卜繁宇)
文献名:Proteomic and N-Terminomic TAILS Analyses of Human Alveolar Bone Proteins: Improved Protein Extr ...
- Journal of Proteome Research | SAAVpedia: identification, functional annotation, and retrieval of single amino acid variants for proteogenomic interpretation | SAAV的识别、功能注释和检索 | (解读人:徐洪凯)
文献名:SAAVpedia: identification, functional annotation, and retrieval of single amino acid variants fo ...
- Journal of Proteome Research | iHPDM: In Silico Human Proteome Digestion Map with Proteolytic Peptide Analysis and Graphical Visualizations(iHPDM: 人类蛋白质组理论酶解图谱的水解肽段分析和可视化展示)| (解读人:邓亚美)
文献名:iHPDM: In Silico Human Proteome Digestion Map with Proteolytic Peptide Analysis and Graphical Vi ...
- Journal of Proteome Research | Down-Regulation of a Male-Specific H3K4 Demethylase, KDM5D, Impairs Cardiomyocyte Differentiation (男性特有的H3K4脱甲基酶基因(KDM5D)下调会损伤心肌细胞分化) | (解读人:徐宁)
文献名:Down-Regulation of a Male-Specific H3K4 Demethylase, KDM5D, Impairs Cardiomyocyte Differentiatio ...
- Journal of Proteome Research | Quantitative Subcellular Proteomics of the Orbitofrontal Cortex of Schizophrenia Patients (精神分裂症病人眶额叶皮层亚细胞结构的定量蛋白质组学研究)(解读人:王聚)
期刊名:Journal of Proteome Research 发表时间:(2019年10月) IF:3.78 单位: 里约热内卢联邦大学 坎皮纳斯州立大学 坎皮纳斯州立大学神经生物学中心 卡拉博大 ...
- Journal of Proteome Research | Proteomic Profiling of Rhabdomyosarcoma-Derived Exosomes Yield Insights into Their Functional Role in Paracrine Signaling (解读人:孙国莹)
文献名:Proteomic Profiling of Rhabdomyosarcoma-Derived Exosomes Yield Insights into Their Functional Ro ...
- Journal of Proteome Research | Global Proteomic Analysis of Lysine Succinylation in Zebrafish (Danio rerio) (解读人:关姣)
文献名:Global Proteomic Analysis of Lysine Succinylation in Zebrafish (Danio rerio)(斑马鱼赖氨酸琥珀酰化的全球蛋白质组学分 ...
随机推荐
- 云服务器之——Linux下配置JDK环境
在Linux下jdk的安装已经操作了很多次,每次发现自己还是会忘记之前的操作,所以今天就简单的来做个记录. 第一步:下载jdk安装包 登录oracle官网:https://www.oracle.com ...
- 获取网站title的脚本
脚本在此 公司的商城需要添加一个脚本,这个脚本就是观察首页页面是否正常,虽然已经配置了zabbix监控网站是否200,但是有一些特殊的情况,比如网页可以打开但是页面是"file not fo ...
- 修改 Cucumber HTML 报告
后台服务是 JSON-RPC 风格的,所以 Scenario 都是这样的 Scenario: login successful When I set request body from "f ...
- iOS多线程之Thread
多线程 • Thread 是苹果官方提供的,简单已用,可以直接操作线程对象.不过需要程序员自己管理线程的生命周期,主要是创建那部分 优缺点 面向对象,简单易用 直接操作线程对象 需要自己管理线程生命周 ...
- 再谈拍照,OPPO这次拿什么和iPhone7拼?
一年一度的iPhone新机如期而至,双摄像头成为iPhone 7 Plus标配,尽管在这之前,双摄像头已有少数厂商在手机上装备,但苹果一出,市场必定全面跟进.无论各大厂商是否采用双摄像头,在手机拍照 ...
- 自制一个可编辑QueryString的类URLModifier
有些情况下,需要 新增/删除/替换 url中的部分Querystring中的参数,而.net自带的Uri类只能解析,不能编辑,,并且如果是Relative类型的链接,转成Uri类型之后,很多参数又不能 ...
- 阿里云ESC学生服务器搭建springboot项目生产环境(Mysql+JDK)不需要上传安装包
嗯,之前服务器被挖矿的病毒弄的登录不进去了,所以联系了阿里云客服,提交工单,最后建议重置,所以我就重置了, 嗯,学习经验,docker如果懂的不是太多,不要随便云部署,都给别人挖矿了. Mysql ...
- Java入门教程十(抽象类接口内部类匿名类)
抽象类(abstract) 一个类只定义了一个为所有子类共享的一般形式,至于细节则交给每一个子类去实现,这种类没有任何具体的实例,只具有一些抽象的概念,那么这样的类称为抽象类. 在面向对象领域,抽象类 ...
- [LeetCode] 207. Course Schedule 课程表
题目: 分析: 这是一道典型的拓扑排序问题.那么何为拓扑排序? 拓扑排序: 有三件事情A,B,C要完成,A随时可以完成,但B和C只有A完成之后才可完成,那么拓扑排序可以为A>B>C或A&g ...
- YiGo表单建立
做一个请假单表单(下图是最后的成品图) 表单的类型 实体表单 1.可存储 2.可编辑 虚拟表单 视图(不可存储数据,只有显示功能) 不可编辑 字典 报表 备注 :一张表单是实体还是虚拟取决于其数据对象 ...