R 绘制 GWAS 研究的 Manhattan 图
曼哈顿图本质上是一个散点图,用于显示大量非零大范围波动数值,最早应用于全基因组关联分析(GWAS)研究展示高度相关位点。它得名源于样式与曼哈顿天际线相似(如下图)。

近几年,在宏基因组领域,尤其是差异OTU结合分类学结果,采用 Manhattan plot 展示有非常好的效果,倍受推崇。
一
曼哈顿图优点
大数据中,即展示数据全貌,又能快速找到目标基因或 OTU,同时可知目标的具体位置和分类、显著程度等信息。绝对高端大气,而且还有内涵。
二
曼哈顿图解读
数据坐标轴介绍,以下图 GWAS 研究结果为例:
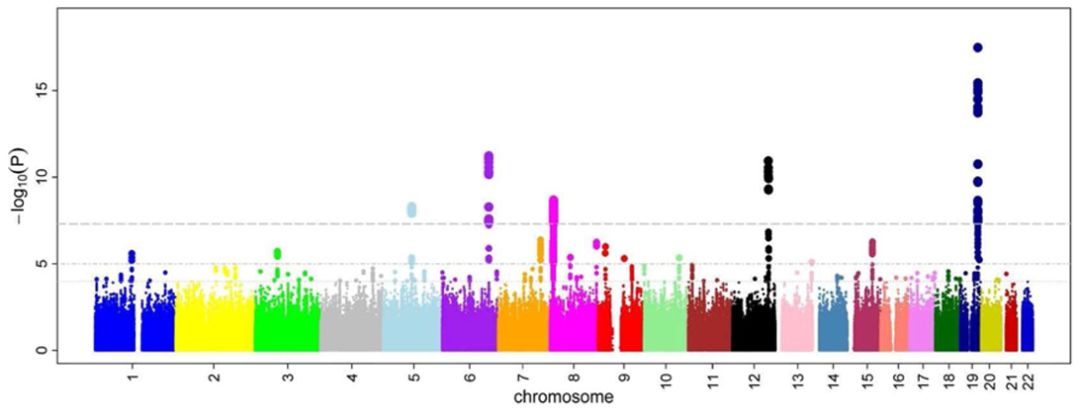
X 轴为染色体编号,且每个基因组 SNP 位点沿染色体序列排列;在 16S 扩增子或宏基因组中则为 OTU 按 Taxonomy 某一级别排序。
Y 轴为该位点相关的统计显著性 Pvalue 值,由于 pvalue 值范围是从 0-1,且越小越好,直接展示非常密集于 0 附近,很难区分。如何使越近 0 的显著数值变大,且而容易区分开,log10 变换是非常好的方法,直接把关注的高显著性(Pvalue 趋近零)值高位显示,远离整体,目标一目了然。
图中水平线一般为设定的不同显著性水平阈值,方便读出每个点的显著性水平;或只添加一条显示性阈值,高于则显著。
三
曼哈顿图绘制工具
R 语言(ggplot2,qqman)、Python(geneview)都能绘制曼哈顿图。本文主要介绍如何使用 R 语言中的 qqman 包绘制 GWAS 研究的 Manhattan 图。
四
R语言 qqman 包
qqman 是一个使用 Q-Q(应用 qq() 函数) 和 manhattan plots(应用 manhattan() 函数) 对 GWAS 分析结果进行可视化的 R 包。
The qqman package includes functions for creating manhattan plots (the manhattan() function) and Q-Q plots (with the qq() function) from GWAS results. The gwasResults data.frame included with the package has simulated results for 16,470 SNPs on 22 chromosomes in a format similar to the output from PLINK.
1. 安装
在 R 中安装 qqman :
# install only once
install.packages("qqman")
# load every time you use it
library(qqman)【左右滑动查看完整信息】
2. 数据
qqman 包中的 gwasResults 数据结构包含了一共 22 条染色体 16470 个 SNP 的 GWAS 模拟结果数据,该数据共4列,分别是:SNP id,染色体编号,SNP 坐标,P value;如下所示(范例的 SNP 坐标是连续的,但真实的 SNP 数据的坐标可能是断续的,但不会影响绘图):
> str(gwasResults)
'data.frame': 16470 obs. of 4 variables:
$ SNP: chr "rs1" "rs2" "rs3" "rs4" ...
$ CHR: int 1 1 1 1 1 1 1 1 1 1 ...
$ BP : int 1 2 3 4 5 6 7 8 9 10 ...
$ P : num 0.915 0.937 0.286 0.83 0.642 ...
> head(gwasResults)
SNP CHR BP P
1 rs1 1 1 0.9148060
2 rs2 1 2 0.9370754
3 rs3 1 3 0.2861395
4 rs4 1 4 0.8304476
5 rs5 1 5 0.6417455
6 rs6 1 6 0.5190959
> tail(gwasResults)
SNP CHR BP P
16465 rs16465 22 530 0.5643702
16466 rs16466 22 531 0.1382863
16467 rs16467 22 532 0.3936999
16468 rs16468 22 533 0.1778749
16469 rs16469 22 534 0.2393020
16470 rs16470 22 535 0.2630441
【左右滑动查看完整信息】
每一条染色体上的 SNPs 数量:
> as.data.frame(table(gwasResults$CHR))
Var1 Freq
1 1 1500
2 2 1191
3 3 1040
4 4 945
5 5 877
6 6 825
7 7 784
8 8 750
9 9 721
10 10 696
11 11 674
12 12 655
13 13 638
14 14 622
15 15 608
16 16 595
17 17 583
18 18 572
19 19 562
20 20 553
21 21 544
22 22 535【左右滑动查看完整信息】
3. 绘制曼哈顿图
绘制最基础曼哈顿图:
manhattan(gwasResults)【左右滑动查看完整信息】
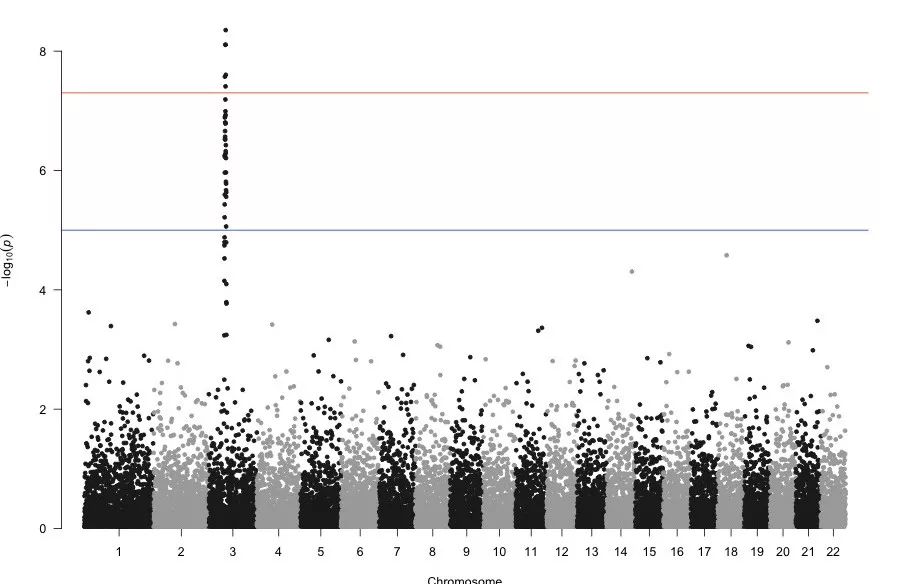
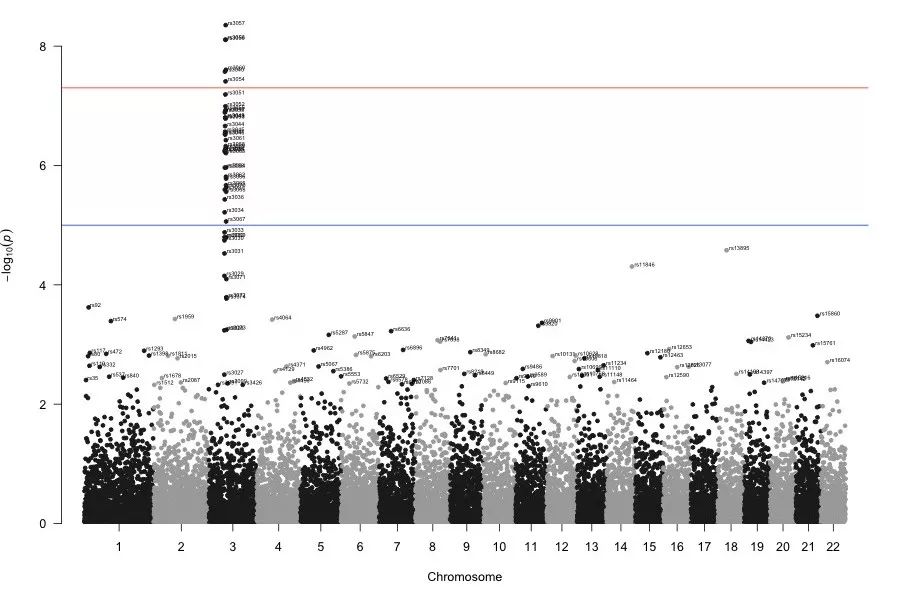
图形说明:
各染色体散点数据来自 gwasResult,是绘图的主数据;
chr3 的线状点来自 qqman 内置的 snpsOfInterest 向量(snpsOfInterest <- paste0("rs", 3001:3100)),由 highlight 参数控制;
灰色横线(suggestive)由参数 suggestiveline 控制,默认 -log10(1e-5) = 5 ;如果为 FALSE,不绘制;
红色横线(genome-wide significance)由参数 genomewideline 控制,默认 -log10(5e-8) ≈ 7.3;如果为 FALSE,不绘制;
灰色、红色横线也可以使用 aline 函数进行绘制:
abline(h=-log10(5e-8), col="red", lty=1, lwd=1)
abline(h=-log10(1e-5), col="blue", lty=1, lwd=1)
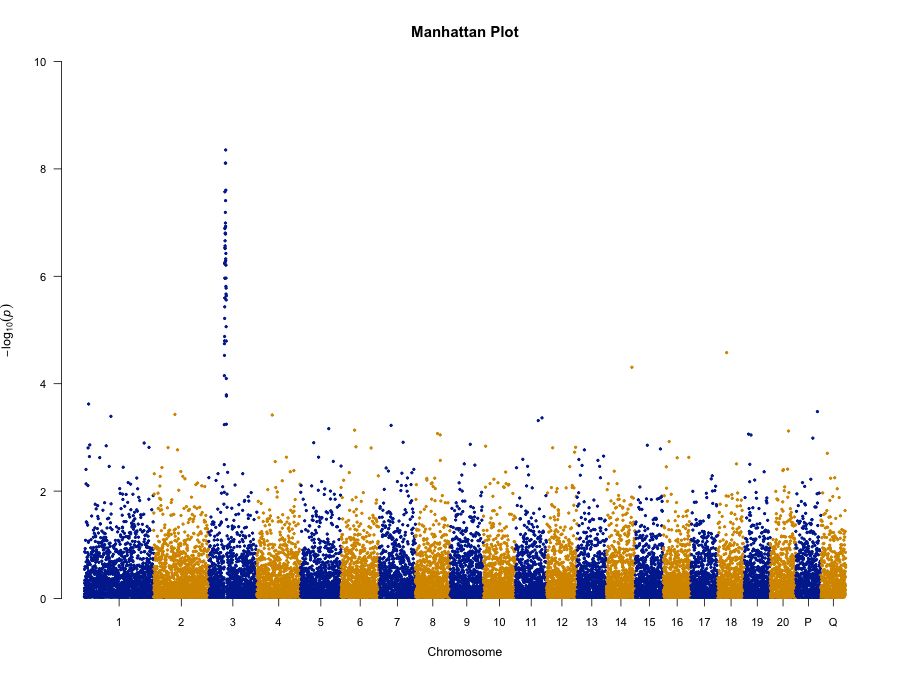
我们可以通过修改参数,更改图形的颜色、标题,以及去除横线:
manhattan(gwasResults,
main = "Manhattan Plot", ## 图形标题
ylim = c(0, 10), ## y 轴绘图范围
cex = 0.6, ## 减少点的大小到 60%
cex.axis = 0.9, ## 减少 X、Y 轴标签的字体大小到 90%
col = c("blue4", "orange3"), ## 散点颜色;该函数会循环利用 col 中的颜色向量
suggestiveline = F, ## 去除 suggestive 参考线
genomewideline = F, ## 去除 genome-wide significance 参考线
chrlabs = c(1:20, "P", "Q")) ## 自定义染色体标号【左右滑动查看完整信息】
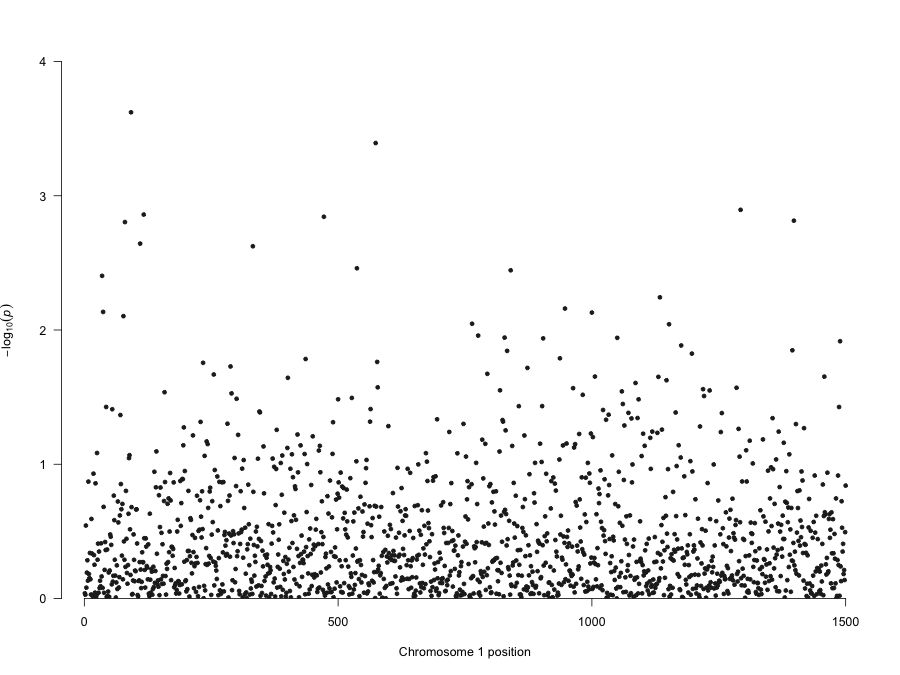
查看单条染色体的分布情况:
manhattan(subset(gwasResults, CHR == 1))【左右滑动查看完整信息】
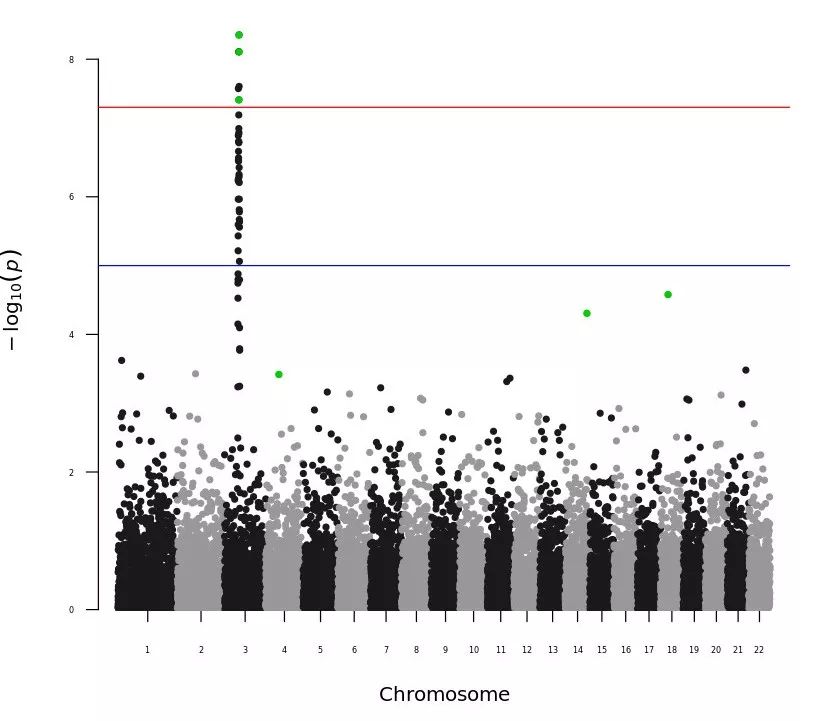
对 chr3 染色体中感兴趣的 SNPs 进行高亮显示。qqman 包内置了一个叫 snpsOfInterest 向量(vector),该向量包含了 100 个位于 chr3 的 SNPs ( snpsOfInterest <- paste0("rs", 3001:3100) )。
如果我们对不存在的 SNPs 进行高亮(highlight)显示,将会引发警告(warning:You're trying to highlight SNPs that don't exist in your results.)。
> str(snpsOfInterest)
chr [1:100] "rs3001" "rs3002" "rs3003" "rs3004" "rs3005" "rs3006" "rs3007" ...
> manhattan(gwasResults, highlight = snpsOfInterest)【左右滑动查看完整信息】
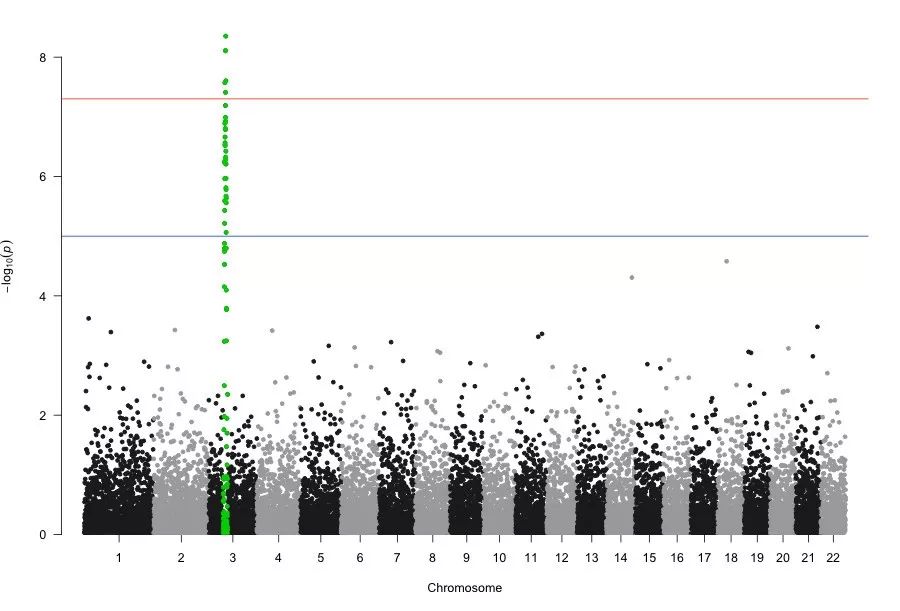
我们也可以自定义感兴趣的 SNPs 进行高亮(highlight)显示,在 GWAS 中如何选择感兴趣的 SNPs:一般是我们做完 gwas 分析,哪个 peak 是你感兴趣的,我们就可以把该 peak 相关的 SNPs 单独提取出来。
> hig = c("rs3057", "rs3056", 'rs3054', "rs4064", "rs11846", "rs13895")
> manhattan(gwasResults, highlight = hig)
#以文件的形式读取感兴趣 SNPs
$ cat hig.txt
rs3057
rs3056
rs3054
rs4064
rs11846
rs13895
> hi = read.table("hig.txt")
> manhattan(gwasResults, highlight = hi$V1)【左右滑动查看完整信息】
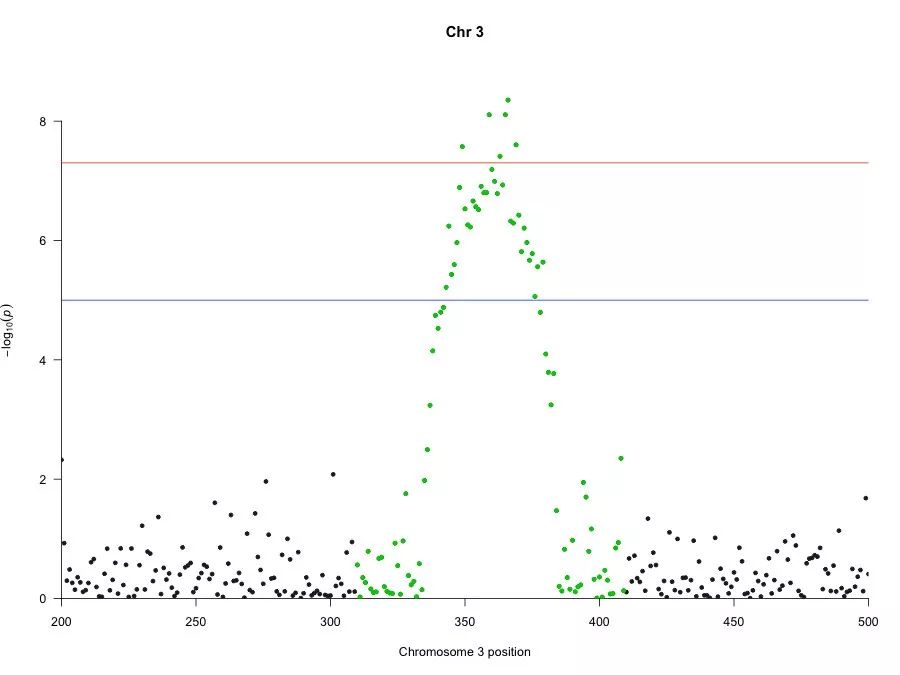
综合高亮和区域限制,我们利用 xlim 参数对位于 chr3:200-500 区域 SNPs 中的 snpsOfInterest 进行高亮显示:
manhattan(subset(gwasResults, CHR == 3), highlight = snpsOfInterest, xlim = c(200, 500), main = "Chr 3")【左右滑动查看完整信息】
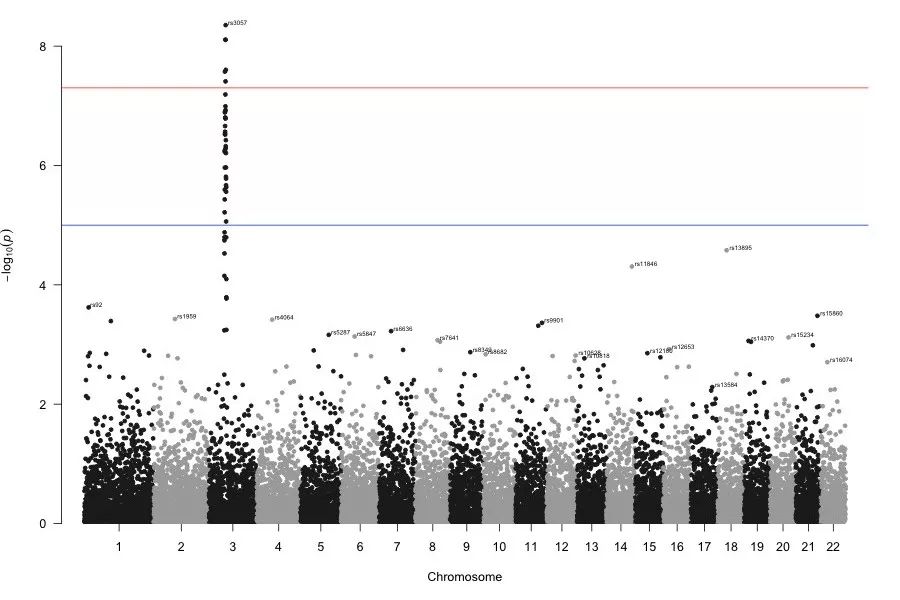
我们可以利用 annotatePval 参数根据 p-value 值对 SNPs 进行注释。该参数默认会对位于每条染色体上,且大于其设定阈值的 Top SNP 进行注释(显示名称):
# -log10(0.001) = 3
manhattan(gwasResults, annotatePval = 0.01)【左右滑动查看完整信息】
我们也可以利用 annotatePval+annotateTop 对符合阈值范围内的所有 SNPs 进行注释:
# -log10(0.005) ≈ 2.3
manhattan(gwasResults, annotatePval = 0.005, annotateTop = FALSE)【左右滑动查看完整信息】
4. 注意事项
通过执行 str(gwasResults),可以看到 gwasResults 数据框包含了 SNP、染色体、位置,以及 p-value 4 列信息,并分别对应命名为 SNP, CHR, BP,以及 P。如果我们使用 qqman 绘制曼哈顿图但输入数据的列名不一样时,需要使用 chr="输入数据染色体列名", bp="输入数据位置列名",p="输入数据 p-value 列名",以及 snp="输入数据 snp 列名" 进行重新设置。
输入数据的染色体(chromosome)列必须为数值(numeric)。如果名字包含 "X", "Y", 或者 "MT",我们必须把它们重命名为 23, 24, 25 等。我们也可以通过修改源码(e.g., fix(manhattan))的方式更改指定轴刻度标签(labs <- unique(d$CHR))为我们想要的任何形式。
如果想要更改高亮(highlight)或者 suggestive/genomewide 线的颜色,我们需要在源码中找到 col="blue"(suggestive line), col="red"(genomewide line), 以及 col="green3"(highlight colors)后分别进行修改。suggestive/genomewide 线的颜色与样式,也可以使用 aline 函数进行修改(参上上文的"图形说明")。
关于 R 绘制 GWAS 研究的 Manhattan 图就介绍到这里,如果你觉得本文章对你有用,欢迎转发!
本文分享自微信公众号 - 生信科技爱好者(bioitee)。
如有侵权,请联系 support@oschina.cn 删除。
本文参与“OSC源创计划”,欢迎正在阅读的你也加入,一起分享。
R 绘制 GWAS 研究的 Manhattan 图的更多相关文章
- ggplot2|玩转Manhattan图-你有被要求这么画吗?
本文首发于“生信补给站”,ggplot2|玩转Manhattan图-你有被要求这么画吗?更多关于R语言,ggplot2绘图,生信分析的内容,敬请关注小号. Manhattan图算是GWAS分析的标配图 ...
- R绘制中国地图,并展示流行病学数据
流行病学的数据讲究“三间分布”,即人群分布.时间分布和空间分布.其中的“空间分布”最好是在地图上展示,才比较清楚.R软件集统计分析与高级绘图于大成,是最适合做这项工作了.关于地图的绘制过程,谢益辉.邱 ...
- 绘制3D的托卡马克位形图的matlab脚本文件 ThreeD.m
% 绘制3D的托卡马克位形图, (V 0.1 by Jiale Chan for Y. H. Huang) % Dee Formula % 特征参数 rzero = 2.0; rmax ...
- GWAS研究中case和control的比例是有讲究的?
GWAS研究中,表型分两种.第一种是线性的表型,如果身高.体重.智力等:第二种是二元的表型,比如患病和未患病,即通常所说的case和control.对于表型是线性的样本来说,是不存在case和cont ...
- 用R包中heatmap画热图
一:导入R包及需要画热图的数据 library(pheatmap) data<- read.table("F:/R练习/R测试数据/heatmapdata.txt",head ...
- 用HTML5绘制的一个星空特效图
<!doctype html> <html lang="en"> <head> <meta charset="UTF-8&quo ...
- 用Python 绘制分布(折线)图
用Python 绘制分布(折线)图,使用的是 plot()函数. 一个简单的例子: # encoding=utf-8 import matplotlib.pyplot as plt from pyla ...
- Unity绘制圆和缓动雷达图
Unity绘制圆和缓动雷达图 之前在做UI模块的时候遇到过需要做雷达图的效果,所以简单复习了一下 关于网格绘制 我们都知道Unity绘制图形是通过Mesh网格添加顶点进行绘制,那么知道顶点信息后如何实 ...
- C#绘制立体三维饼状图
转载自原文 C#绘制立体三维饼状图(超酷) 内容原文地址:LINK [翻译]Julijan Sribar著3D Pie Chart一个用于绘制立体三维饼状图的C#类库[简介]本文的想法就是创建一个独立 ...
- 使用highcharts绘制美观的燃尽图
使用highcharts绘制美观的燃尽图 助教在博客中介绍了两种绘制燃尽图的方法,但是我们组在使用时发现有些任务不适合写进issue,而且网站生成的燃尽图不是很美观,因此我们打算使用其他方法自己绘制燃 ...
随机推荐
- 这样封装echarts简单好用
为什么要去封装echarts? 在我们的项目中,有很多的地方都使用了echarts图表展示数据. 在有些场景,一个页面有十多个的echarts图. 这些echarts只是展示的指标不一样. 如果我们每 ...
- springboot实现验证码功能
实现验证码功能 先在utils包下创建一个ValidateImageCodeUtils.class package com.wfszmg.demo.utils; import javax.imagei ...
- CAS乐观锁(原子操作)
更多内容,前往 IT-BLOG 锁主要分为两种:乐观锁和悲观锁,而 synchronized 就属于一种悲观锁,每次在操作数据前都会加锁.乐观锁是指:乐观的认为自己在操作数据时,别人不会对当前数据进行 ...
- Barplot/pie/boxplot作图详解——R语言
当数据以简单的可视化的形式呈现时,数据便更具有意义并且更容易理解,因为人眼很难从原始数据中得出重要的信息.因此,数据可视化成为了解读数据最重要的方式之一.条形图和箱线图是了解变量分布的最常用的图形工具 ...
- mysql的concat与concat_ws拼接字符串的使用
concat的使用 可以拼接多个字符 mysql> select concat(name,dept,job) from t1; +-----------------------+ | conca ...
- GitLab 安装部署使用
GitLab介绍 GitLab:是一个基于Git实现的在线代码仓库托管软件,你可以用gitlab自己搭建一个类似于Github一样的系统,一般用于在企业.学校等内部网络搭建git私服. 功能:Gitl ...
- Bootstrapd导航条使用
要想在程序中集成Bootstrap,显然要对模板做所有必要的改动.不过,更简单的方法是使用一个名为Flask-Bootstrap 的Flask 扩展,简化集成的过程. 安装:Flask-Bootstr ...
- python入门教程之一 什么是python
python简介 1 什么是python Python是一种计算机程序设计语言.你可能已经听说过很多种流行的编程语言,比如非常难学的C语言,非常流行的Java语言,适合初学者的Basic语言,适合网页 ...
- 如何训练个人的ChatGpt4
如何在自己的计算机上安装类似 ChatGPT 的个人 AI 并在没有互联网的情况下运行它 本文旨在为任何人安装此软件.最初它有一个视频,伴随着操作方法,但是事情变化很快,我的三次尝试只是推迟了我发表这 ...
- Docker 配置阿里云或腾讯云镜像加速
1.新建 /etc/docker/daemon.json 文件,并写入以下内容: 阿里云按下面配置 sudo tee /etc/docker/daemon.json <<-'EOF' { ...