DESeq2包
1)简介:
DESeq2-package: for differential analysis of count data(对count data 做差异分析)
2)安装
if("DESeq2" %in% rownames(installed.packages()) == FALSE) {source("http://bioconductor.org/biocLite.R");biocLite("DESeq2")}
suppressMessages(library(DESeq2))
ls('package:DESeq2')
3)对象的使用说明
3.1)coef(Extract a matrix of model coefficients/standard errors,高级用户检验模型系数)
语法:coef(object, SE = FALSE, ...)
参数解释:
object:a DESeqDataSet returned by DESeq, nbinomWaldTest, or nbinomLRT.
例子:
dds <- makeExampleDESeqDataSet(m=4)
dds <- DESeq(dds)
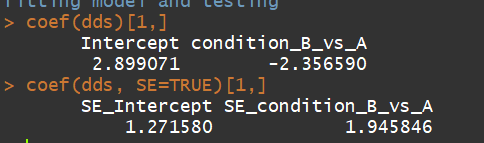
coef(dds)[1,]
coef(dds, SE=TRUE)[1,]
3.2) collapseReplicates:Collapse technical replicates in a RangedSummarizedExperiment or DESeqDataSet(用于消除技术重复)
用法:collapseReplicates(object, groupby, run, renameCols = TRUE)
参数:
object:A RangedSummarizedExperiment or DESeqDataSet
groupby:a grouping factor, as long as the columns of object,分组因子
run:optional, the names of each unique column in object. if provided, a new column runsCollapsed will be added to the colData which pastes together the names of run (测序run)
renameCols:whether to rename the columns of the returned object using the levels of the grouping factor
例子:
dds <- makeExampleDESeqDataSet(m=12)
str(dds)
dds$sample <- factor(sample(paste0("sample",rep(1:9, c(2,1,1,2,1,1,2,1,1))))) (#共9个样品:其中 3个样品有2个技术重重)
dds$run <- paste0("run",1:12) #12个run道
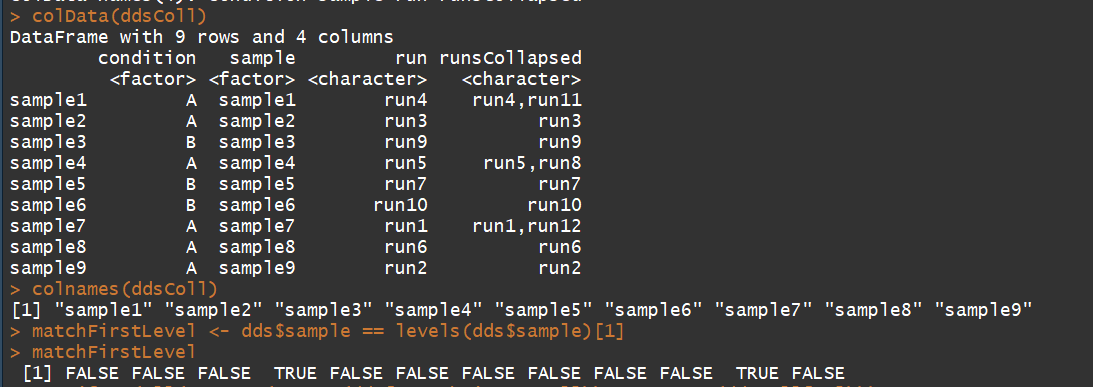
ddsColl <- collapseReplicates(dds, dds$sample, dds$run)
# examine the colData and column names of the collapsed data
colData(ddsColl)
colnames(ddsColl)
# check that the sum of the counts for "sample1" is the same
# as the counts in the "sample1" column in ddsColl
matchFirstLevel <- dds$sample == levels(dds$sample)[1]
stopifnot(all(rowSums(counts(dds[,matchFirstLevel])) == counts(ddsColl[,1])))
3.3)counts:Accessors for the ’counts’ slot of a DESeqDataSet object(对表达矩阵进行统计,)
one row for each observational unit (gene or the like), and one column for each sample(行代表观察值(例如基因),列代表样本(例如肝、脾、肾等))
语法:counts(object, normalized = FALSE,replaced = FALSE)
参数:
object:a DESeqDataSet object(表达矩阵).
normalized:logical indicating whether or not to divide the counts by the size factors or normalization factors before returning (normalization factors always preempt size factors),(即不同量级的数据要不要归一化)
replaced:返回极端值
dds <- makeExampleDESeqDataSet(m=4) ##构建一个表达矩阵
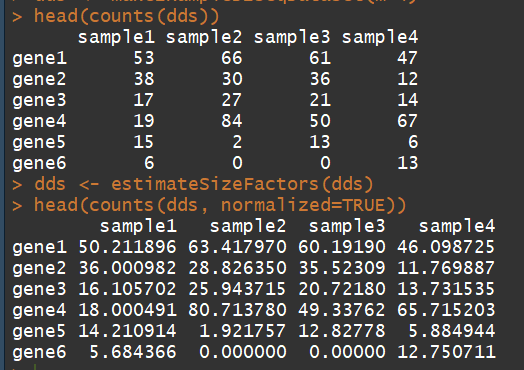
head(counts(dds))
dds <- estimateSizeFactors(dds) # run this or DESeq() first
head(counts(dds, normalized=TRUE))
3.4)DESeq:Differential expression analysis based on the Negative Binomial (a.k.a.Gamma-Poisson) distribution(基于负二项分布进行差异分析)
语法:
DESeq(object, test = c("Wald", "LRT"), fitType = c("parametric", "local","mean"), sfType = c("ratio", "poscounts", "iterate"), betaPrior,full = design(object), reduced, quiet = FALSE,minReplicatesForReplace = 7, modelMatrixType, useT = FALSE, minmu = 0.5,
parallel = FALSE, BPPARAM = bpparam())
参数:
object:a DESeqDataSet object(表达矩阵对象)
test:Wald" or "LRT"检验
fitType:either "parametric", "local", or "mean"
sfType:either "ratio", "poscounts", or "iterate" for teh type of size factor estimation.
betaPrior:whether or not to put a zero-mean normal prior on the non-intercept coefficients
reduced:for test="LRT", a reduced formula to compare against
quiet:whether to print messages at each step
minReplicatesForReplace:the minimum number of replicates required
modelMatrixType:either "standard" or "expanded", which describe how the model matrix, X of the GLM formula is formed.
useT:logical, passed to nbinomWaldTest, default is FALSE
minmu:lower bound on the estimated count for fitting gene-wise dispersion
parallel:if FALSE, no parallelization. if TRUE, parallel execution using BiocParallel,
BPPARAM:an optional parameter object passed internally to bplapply when parallel=TRUE.
例子:
# count tables from RNA-Seq data
cnts <- matrix(rnbinom(n=1000, mu=100, size=1/0.5), ncol=10)
cond <- factor(rep(1:2, each=5)) # object construction
dds <- DESeqDataSetFromMatrix(cnts, DataFrame(cond), ~ cond) # standard analysis
dds <- DESeq(dds)
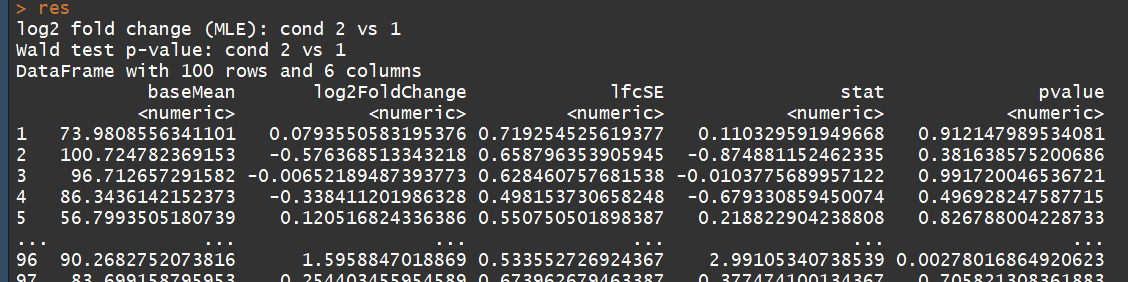
res <- results(dds) # moderated log2 fold changes
resultsNames(dds)
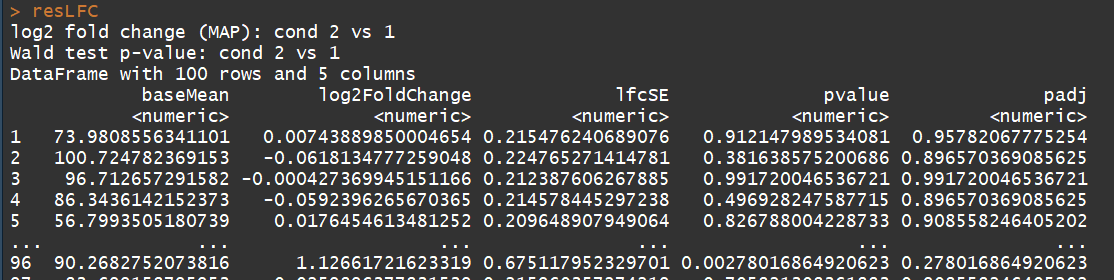
resLFC <- lfcShrink(dds, coef=2, type="apeglm") # an alternate analysis: likelihood ratio test
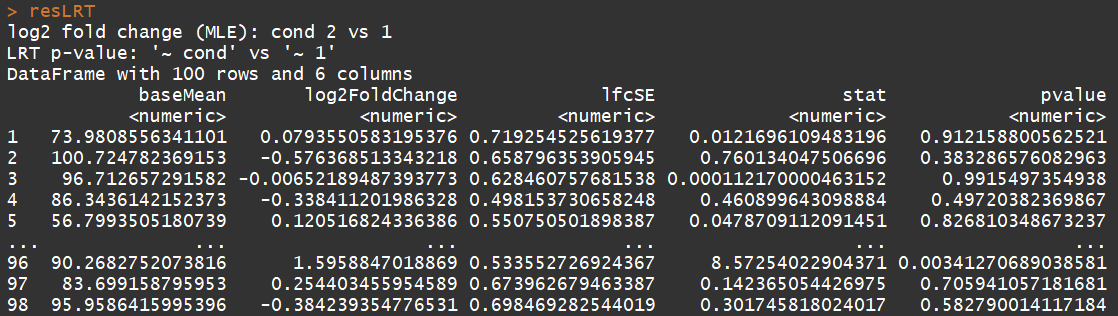
ddsLRT <- DESeq(dds, test="LRT", reduced= ~ 1)
resLRT <- results(ddsLRT)
3.5)DESeqDataSet-class(DESeqDataSet object and constructors)
语法:
DESeqDataSet(se, design, ignoreRank = FALSE)
DESeqDataSetFromMatrix(countData, colData, design, tidy = FALSE,ignoreRank = FALSE, ...)
DESeqDataSetFromHTSeqCount(sampleTable, directory = ".", design,ignoreRank = FALSE, ...)
DESeqDataSetFromTximport(txi, colData, design, ...)
例子:
countData <- matrix(1:100,ncol=4)
condition <- factor(c("A","A","B","B"))
dds <- DESeqDataSetFromMatrix(countData, DataFrame(condition), ~ condition)
3.6)DESeqResults-class:DESeqResults object and constructor
语法:DESeqResults(DataFrame, priorInfo = list())
参数:
DataFrame:a DataFrame of results, standard column names are: baseMean, log2FoldChange,lfcSE, stat, pvalue, padj.
priorInfo:a list giving information on the log fold change prior
3.7)DESeqTransform-class(DESeqTransform object and constructor)
语法:DESeqTransform(SummarizedExperiment)
参数:SummarizedExperiment a RangedSummarizedExperiment
3.8)rlog Apply a ’regularized log’ transformation
用法:
rlog(object, blind = TRUE, intercept, betaPriorVar, fitType = "parametric")
rlogTransformation(object, blind = TRUE, intercept, betaPriorVar,fitType = "parametric")
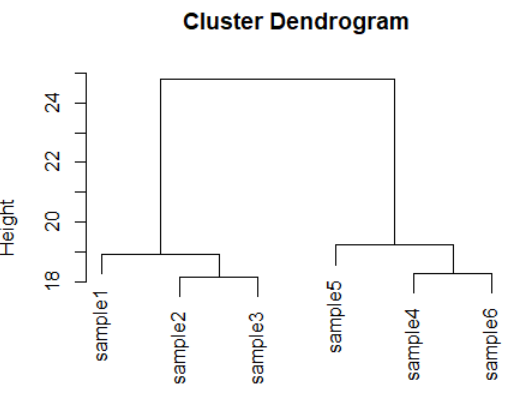
dds <- makeExampleDESeqDataSet(m=6,betaSD=1)
rld <- rlog(dds)
dists <- dist(t(assay(rld)))
plot(hclust(dists))
3.9)plotPCA(Sample PCA plot for transformed data)
用法:plotPCA(object, intgroup = "condition",ntop = 500, returnData = FALSE)
参数:
object:a DESeqTransform object, with data in assay(x), produced for example by either rlog or varianceStabilizingTransformation.
intgroup: interesting groups: a character vector of names in colData(x) to use for grouping
ntop:number of top genes to use for principal components, selected by highest row variance
returnData:should the function only return the data.frame of PC1 and PC2 with intgroup covariates for custom plotting
# using rlog transformed data:
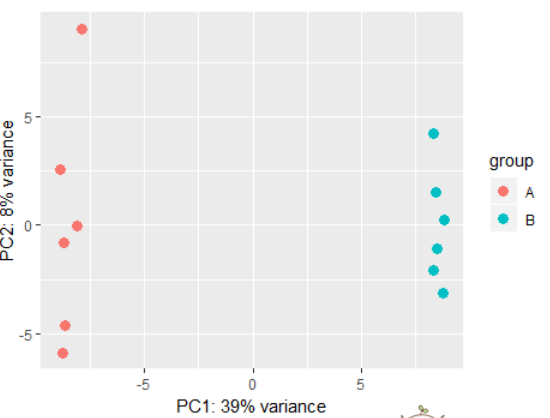
dds <- makeExampleDESeqDataSet(betaSD=1)
rld <- rlog(dds)
plotPCA(rld) # also possible to perform custom transformation:
dds <- estimateSizeFactors(dds)
# shifted log of normalized counts
se <- SummarizedExperiment(log2(counts(dds, normalized=TRUE) + 1),
colData=colData(dds))
# the call to DESeqTransform() is needed to
# trigger our plotPCA method.
plotPCA( DESeqTransform( se ) )
3.10)
DESeq2包的更多相关文章
- 简单使用DESeq2/EdgeR做差异分析
简单使用DESeq2/EdgeR做差异分析 Posted: 五月 07, 2017 Under: Transcriptomics By Kai no Comments DESeq2和EdgeR都 ...
- airway之workflow
1)airway简介 在该workflow中,所用的数据集来自RNA-seq,气道平滑肌细胞(airway smooth muscle cells )用氟美松(糖皮质激素,抗炎药)处理.例如,哮喘患 ...
- miRAN 分析以及mRNA分析
一些参考资料 http://www.360doc.com/content/17/0528/22/19913717_658086490.shtml https://www.cnblogs.com/tri ...
- Error in library(DESeq2) : 不存在叫‘DESeq2’这个名字的程辑包
Error in read.dcf(file.path(pkgname, "DESCRIPTION"), c("Package", "Type&quo ...
- DESeq2 install --- 如何安装R包("RcppArmadillo")?
安装R包("RcppArmadillo")失败,导致依赖该包的DESeq2 无法使用: 首先对gcc,g++升级至4.7, 但依然报错,还是安装不了RcppArmadillo: 报 ...
- R包安装的正确方式
options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/")) if(! req ...
- Npm包的开发
个人开发包的目录结构 ├── coverage //istanbul测试覆盖率生成的文件 ├── index.js //入口文件 ├── introduce.md //说明文件 ├── lib │ ...
- Windows server 2012 添加中文语言包(英文转为中文)(离线)
Windows server 2012 添加中文语言包(英文转为中文)(离线) 相关资料: 公司环境:亚马孙aws虚拟机 英文版Windows2012 中文SQL Server2012安装包,需要安装 ...
- 如何在nuget上传自己的包+搭建自己公司的NuGet服务器(新方法)
运维相关:http://www.cnblogs.com/dunitian/p/4822808.html#iis 先注册一个nuget账号https://www.nuget.org/ 下载并安装一下Nu ...
随机推荐
- java web 程序---投票系统
1.这里会连接数据库--JDBC的学习实例 一共有3个页面. 2.第一个页面是一个form表单,第二个页面是处理数据,第三个页面是显示页面 vote.jsp <body bgcolor=&quo ...
- Linux期中架构 全网备份案例
server端脚本 #!/bin/bash #1 进行数据完整性验证 并生成结果 find /backup -type f -name "finger.txt"| xargs md ...
- win7连接centos的nfs
nfs的服务端这边设置忽略. 主要关于权限的问题: Windows7 NFS客户端,使用mount命令挂载NFS服务之后,文件系统对Win7只读,无法写入文件,无法新建文件夹解决方法. 在win的cm ...
- 手贱,写个call玩玩.
今天在睡觉醒时,突然感觉对call和apply有了点理解,但是又不好表达出来.就随便写几个例子. function say() { return this.role; } function Fathe ...
- 第1课 学习 C++ 的意义
1. 回顾历史 (1)UNIX操作系统诞生之初是直接用汇编语言写成的.随着UNIX的发展,汇编语言的开发效率成为一个瓶劲. (2)1971年,Ken Thompson和Denis Ritchie对B ...
- unity3d之Editor的Assembly-CSharp.dll文件路径
在Editor中与自己project中使用的Mono与Managed文件夹路径区别: Editor中:在unity安装路径[AppDir]下: 自己project中:在project的路径下,由bui ...
- php 编程笔记分享
php获取POST数据的三种方法php 图片加水印源代码php+ajax+json的一个最简单实例php 汉字转拼音源码php遍历目录,生成目录下每个文件的md5值并写入到结果文件中php实现linu ...
- spark 应用程序部署工具 spark-submit
打包 Spark application 使用spark-submit启动Spark application spark-submit usage spark-submit option 运行模式相关 ...
- 深度强化学习——连续动作控制DDPG、NAF
一.存在的问题 DQN是一个面向离散控制的算法,即输出的动作是离散的.对应到Atari 游戏中,只需要几个离散的键盘或手柄按键进行控制. 然而在实际中,控制问题则是连续的,高维的,比如一个具有6个关节 ...
- sql上下级关系查询
有一张存在上下级关系的function表,parentId表示上级Id,现要查询出上级菜单下的子菜单,每个子菜单显示成以逗号分隔的字符串 表结构如下: create table menu ( id i ...