Bioconductor 中的 R 包安装教程
Bioconductor 是一个基于 R 语言的生物信息软件包,主要用于生物数据的注释、分析、统计、以及可视化(http://www.bioconductor.org)。
总所周知,Bioconductor 是和 R 版本绑定的,这是为了确保用户不把包安装在错误的版本上。Bioconductor 发行版每年更新两次,它在任何时候都有一个发行版本(release version),对应于 R 的发行版本。此外,Bioconductor 还有一个开发版本(development version),它对应于 R 的开发版本。
R 每年(通常是 4 月中旬)在 ‘x.y.z’ 中发布一个 ‘.y’ 版本,但 Bioconductor 每 6 个月(4 月中旬和 10 月中旬)发布一个 ‘.y’ 版本。
Bioconductor 与 R 各自对应的版本如下:(Bioconductor releases) 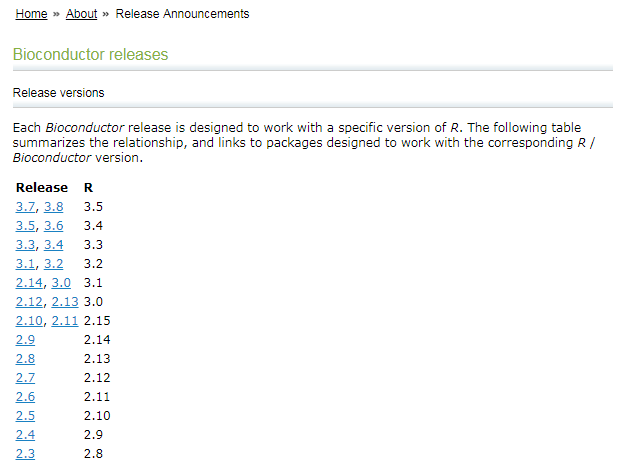
biocLite
在 R==3.5(Bioconductor-3.7 前,Bioconductor 都是通过 biocLite 安装相关的 R 包:
source("https://bioconductor.org/biocLite.R")
biocLite(pkg_name)但是,从 R-3.5(Bioconductor-3.8)起,Bioconductor 更改了 R 包的安装方式:它们通过发布在 CRAN 的 BiocManager 包来对 Bioconductor 的包进行安装和管理——通过 CRAN 安装 BiocManager,再通过这个包来安装 Bioconductor 的包。
BiocManager
安装 BiocManager 包
chooseCRANmirror()
install.packages("BiocManager")
安装 Bioconductor (or CRAN) 的 R 包
BiocManager::install(c("GenomicRanges", "Organism.dplyr"))
更新所有已经安装的 R 包到最新版本
BiocManager::install()
查看 Bioconductor 的版本
BiocManager::version()
## '3.8'
旧和意外版本的 R 包
当 Bioconductor 的包都来自同一版本时,它们的效果最佳。 使用 valid() 来查看过期(out-of-date)或意外版本(unexpected versions)的 R 包。
BiocManager::valid()
## Warning: 21 packages out-of-date; 2 packages too new
##
## * sessionInfo()
##
## R Under development (unstable) (2018-11-02 r75540)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 18.04.1 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.7.1
## LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.7.1
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=C
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] BiocStyle_2.11.0
##
## loaded via a namespace (and not attached):
## [1] Rcpp_1.0.0 bookdown_0.7 digest_0.6.18
## [4] rprojroot_1.3-2 backports_1.1.2 magrittr_1.5
## [7] evaluate_0.12 stringi_1.2.4 rmarkdown_1.10
## [10] tools_3.6.0 stringr_1.3.1 xfun_0.4
## [13] yaml_2.2.0 compiler_3.6.0 BiocManager_1.30.4
## [16] htmltools_0.3.6 knitr_1.20
##
## Bioconductor version '3.9'
##
## * 21 packages out-of-date
## * 2 packages too new
##
## create a valid installation with
##
## BiocManager::install(c(
## "BiocManager", "GenomicDataCommons", "GenomicRanges", "IRanges",
## "RJSONIO", "RcppArmadillo", "S4Vectors", "TCGAbiolinks", "TCGAutils",
## "TMB", "biocViews", "biomaRt", "bumphunter", "curatedMetagenomicData",
## "dimRed", "dplyr", "flowCore", "ggpubr", "ggtree", "lme4", "rcmdcheck",
## "shinyFiles", "tximportData"
## ), update = TRUE, ask = FALSE)
##
## more details: BiocManager::valid()$too_new, BiocManager::valid()$out_of_datevalid() 返回一个对象,可以查询该对象以获取有关无效包的详细信息:
> v <- valid()
Warning message:
6 packages out-of-date; 0 packages too new
> names(v)
[1] "out_of_date" "too_new"
> head(v$out_of_date, 2)
Package LibPath
bit "bit" "/home/shenweiyan/R/x86_64-pc-linux-gnu-library/3.5-Bioc-3.8"
ff "ff" "/home/shenweiyan/R/x86_64-pc-linux-gnu-library/3.5-Bioc-3.8"
Installed Built ReposVer Repository
bit "1.1-12" "3.5.0" "1.1-13" "https://cran.rstudio.com/src/contrib"
ff "2.2-13" "3.5.0" "2.2-14" "https://cran.rstudio.com/src/contrib"
>
适用的 R 包
可以使用 available() 发现适用于我们的 Bioconductor 版本的软件包; 第一个参数是可用于根据正则表达式过滤包名称,例如,可用于 Homo sapiens 的 ‘BSgenome’ 包:
avail <- BiocManager::available()
length(avail)
## [1] 16261
BiocManager::available("BSgenome.Hsapiens")
## [1] "BSgenome.Hsapiens.1000genomes.hs37d5"
## [2] "BSgenome.Hsapiens.NCBI.GRCh38"
## [3] "BSgenome.Hsapiens.UCSC.hg17"
## [4] "BSgenome.Hsapiens.UCSC.hg17.masked"
## [5] "BSgenome.Hsapiens.UCSC.hg18"
## [6] "BSgenome.Hsapiens.UCSC.hg18.masked"
## [7] "BSgenome.Hsapiens.UCSC.hg19"
## [8] "BSgenome.Hsapiens.UCSC.hg19.masked"
## [9] "BSgenome.Hsapiens.UCSC.hg38"
## [10] "BSgenome.Hsapiens.UCSC.hg38.masked"Bioconductor 中安装旧版本的 R 包
对于 R >= 3.5,Bioconductor-3.8 可以使用 BiocManager 安装相关与版本匹配的 R 包。那么使用 3.5 以下 R 版本的用户是继续使用 biocLite,还是 BiocManager,还是其他的方法安装匹配相关版本的 R 包呢?
首先,biocLite 和 BiocManager 不适用 R < 3.5。
> source("https://bioconductor.org/biocLite.R")
Bioconductor version 3.6 (BiocInstaller 1.28.0), ?biocLite for help
A new version of Bioconductor is available after installing the most recent
version of R; see http://bioconductor.org/install
> biocLite("clusterProfile")
......
Warning message:
package ‘clusterProfile’ is not available (for R version 3.4.3)
> chooseCRANmirror()
> install.packages("BiocManager")
Warning message:
package ‘BiocManager’ is not available (for R version 3.4.3)
>其次,对于 R < 3.5.0,Bioconductor 推荐使用以下命令安装相应的 R 包。
source("https://bioconductor.org/biocLite.R")
BiocInstaller::biocLite(c("GenomicFeatures", "AnnotationDbi"))Bioconductor 安装旧版本 clusterProfiler
在 Aanconda2 环境 R==3.4.3 中安装 clusterProfiler,发现 package ‘clusterProfile’ is not available (for R version 3.4.3)。
> source("https://bioconductor.org/biocLite.R")
Bioconductor version 3.6 (BiocInstaller 1.28.0), ?biocLite for help
A new version of Bioconductor is available after installing the most recent
version of R; see http://bioconductor.org/install
> biocLite("clusterProfile")
BioC_mirror: https://bioconductor.org
Using Bioconductor 3.6 (BiocInstaller 1.28.0), R 3.4.3 (2017-11-30).
Installing package(s) ‘clusterProfile’
Old packages: 'ade4', 'ape', 'backports', 'caret', ......
Update all/some/none? [a/s/n]: n
Warning message:
package ‘clusterProfile’ is not available (for R version 3.4.3)使用 BiocInstaller 安装 clusterProfiler:
> source("https://bioconductor.org/biocLite.R")
Bioconductor version 3.6 (BiocInstaller 1.28.0), ?biocLite for help
A new version of Bioconductor is available after installing the most recent
version of R; see http://bioconductor.org/install
> BiocInstaller::biocLite("clusterProfiler")
BioC_mirror: https://bioconductor.org
Using Bioconductor 3.6 (BiocInstaller 1.28.0), R 3.4.3 (2017-11-30).
Installing package(s) ‘clusterProfiler’
trying URL 'https://bioconductor.org/packages/3.6/bioc/src/contrib/clusterProfiler_3.6.0.tar.gz'
Content type 'application/x-gzip' length 4478098 bytes (4.3 MB)
==================================================
downloaded 4.3 MB
* installing *source* package ‘clusterProfiler’ ...
** R
** data
** inst
** byte-compile and prepare package for lazy loading
** help
*** installing help indices
** building package indices
** installing vignettes
** testing if installed package can be loaded
* DONE (clusterProfiler)
> packageVersion('clusterProfiler')
[1] ‘3.6.0’
本文分享自微信公众号 - 生信科技爱好者(bioitee)。
如有侵权,请联系 support@oschina.cn 删除。
本文参与“OSC源创计划”,欢迎正在阅读的你也加入,一起分享。
Bioconductor 中的 R 包安装教程的更多相关文章
- R 包安装、载入和卸载
生物上的一些包可以这样安装 source("https://bioconductor.org/biocLite.R") biocLite("affy") 一般的 ...
- Linux环境下R和R包安装及其管理
前言 R对windows使用很友好,对Linux来说充满了敌意.小数据可以在windows下交互操作,效果很好很棒.可是当我们要处理大数据,或者要在集群上搭建pipeline时,不得不面对在Linux ...
- Sublime Text 2安装汉化破解、插件包安装教程
原文地址: Sublime Text 2安装汉化破解.插件包安装教程_百度经验 http://jingyan.baidu.com/article/ff4116259b057c12e48237b8.ht ...
- windows系统mysql-5.7官方绿色版zip包安装教程
准备 下载页面:https://dev.mysql.com/downloads/mysql/ 点击 Download 按钮下载zip包到本地,解压(以我本地的解压路径是 D:\db\mysql-5.7 ...
- 如何将R包安装到自定义路径
参考 设置环境变量R_LIBS将R包安装到自定义路径 实际上是可以解决问题的, #环境变量完成以后,启动(重启)R,运行 .libPaths() 加载R包时,发现路径仍然未变成自定义的. 那么参 ...
- R包安装的正确方式
options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/")) if(! req ...
- Hadoop学习---Ubuntu中hadoop完全分布式安装教程
软件版本 Hadoop版本号:hadoop-2.6.0-cdh5.7.0: VMWare版本号:VMware 9或10 Linux系统:CentOS 6.4-6.5 或Ubuntu版本号:ubuntu ...
- VirtualBox扩展包安装教程|VirtualBox扩展增强包怎么安装
VirtualBox是一款功能强大的免费虚拟机软件,一般我们安装VirtualBox后要安装扩展增强包,VirtualBox扩展包包含USB2.0和USB3.0控制等支持功能,如果没有装,在使用过程中 ...
- vs中python包安装教程
vs安装python很简单,只需要在vs安装包中选择python就可以了,这里使用的python3.7: 如果有了解,都知道安装python包的指令:"pip install xxx&quo ...
- Xbox360自制系统GOD包安装教程
1.准备工作 U盘或移动硬盘一个,已下载好的GOD包,本教程用一个32G的U盘和游戏<猎天使魔女>为例. 右击U盘,属性,查看你的U盘是否为FAT32格式. 如果是FAT32格式,则可直接 ...
随机推荐
- Oracle数据库 insert 插入数据 显示问号乱码的解决办法
一.问题描述 插入的中文数据 显示成问号(乱码),其他语言如老挝文.柬文等都一样. 二.解决方案 plsql插入oracle数据乱码问题处理起来其实很简单,因为乱码问题一般都是由于编码不一致导致的,我 ...
- Mysql 备份方案
一.为什么要备份 [1]容灾恢复:硬件故障.不经意的 Bug 导致数据损坏,或者服务器及其数据由于某些原因不可获取或无法使用等(例如:机房大楼烧毁,恶意的黑客攻击或 Mysql 的 Bug 等).[2 ...
- ML-程序练习-Dragon
回归问题 前期 假设已有某样例,参数为w=1.477, b=0.089,即为\(y=1.477x+0.089\) 过程分析 数据采样 首先我们需要模拟一些带有真实样本观测误差的数据(因为真实情况是真实 ...
- Dijkstra(迪杰斯特拉)算法C++实现&讲解
Dijkstra迪杰斯特拉算法及C++实现 Dijkstra算法是典型的最短路径路由算法,用来计算一个节点到其他所有节点的最短路径.算法的基本思想和流程是:1. 初始化出发点到其它各点的距离dist[ ...
- 彻底弄懂C#中delegate、event、EventHandler、Action、Func的使用和区别
[目录] 1 委托 2 事件-概念的引出 3 事件-关于异常 4 事件-关于异步 5 委托-Func与Action 1 委托 在.NET中定义"委托"需要用到delegate关键字 ...
- vue之数组的方法
目录 简介 filter方法 简介 本文会把遇到的数组的方法慢慢补充进来 filter方法 filter()方法是一个过虑方法 以下面的为例:列表dataList会每次取一个值,把值给匿名函数,并执行 ...
- pysimplegui之使用多线程,避免程序卡死
这个问题我也遇到过,就是还需要一个while循环的时候,放到gui本身循环会卡死,这时候就需要启动多线程 需要"长时间"的操作 如果您是 Windows 用户,您会在其标题栏中看到 ...
- Java设计模式 —— 面向对象设计原则
1 设计模式概述 1.1 设计模式的定义与分类 设计模式的定义 Design patterns are descriptions of communicating objects and classe ...
- 【Spring注解驱动】(二)AOP及一些扩展原理
1 AOP动态代理简介及功能实现 1.1 简介 指在程序运行期间动态地将某段代码切入到指定方法的指定位置进行运行的方式. 1.2 功能实现测试 功能:实现在业务逻辑运行的时候将日志打印 ①导入aop模 ...
- MQTT.fx的安装和使用
一.下载和安装 MQTT.fx支持Windows/Linux/Mac,附下载地址:http://www.jensd.de/apps/mqttfx/,下载完成之后双击进行安装. 二.配置使用 打开软件, ...