sam/bam格式
1)Sam (Sequence Alignment/Map)
-------------------------------------------------
1) SAM 文件产生背景
随着Illumina/Solexa, AB/SOLiD and Roche/454测序技术不断的进步,各种比对工具产生,被用来高效的将reads比对到参考基因组。因为这些比对工具产生不同格式的文件,导致下游分析比较困难,因此一个通用的格式可以提供一个很好的接口用于链接比对与下游分析(组装,变异等,基因分型等)。因此SAM格式应运而生,主要是用来存储测序reads与参考序列比对结果信息的一种文件格式,以TAB为分割符,支持不同平台的短reads及长reads(最长为128Mbp)。
2)格式解读
我们用文献中的例子来详细解释sam格式。
2.1)首先看一个比对事件:

ref是参考序列,Read r001/1和 r001/2组成read pair,r003是嵌合体(chimeric read) ,r004表示 split alignment事件
2.2)相应的sam格式是:
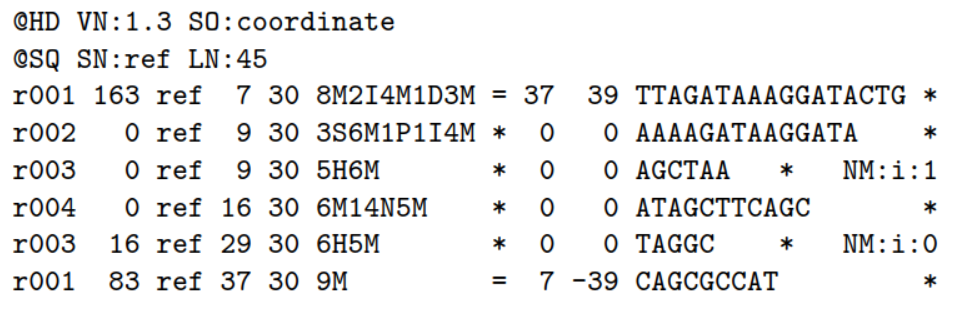
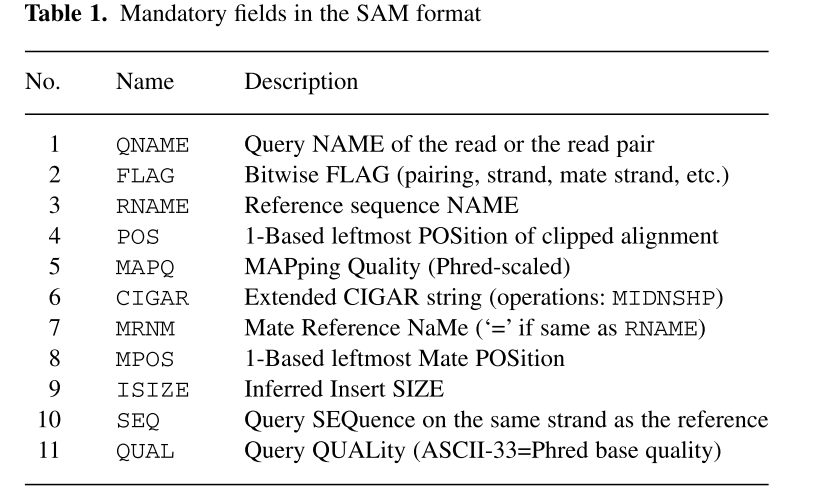
这11列内容的解释:
由此我们可以看到,SAM是由两部分组成:分为标头注释信息(header section)和比对结果(alignment section)。标头信息可有可无,都是以@开头,用不同的tag表示不同的信息,主要有:
@HD,说明符合标准的版本、对比序列的排列顺序(这里为coordinate)
@SQ,参考序列说明 (SN:ref,LN 是参考序列的长度)
@PG,使用的比对程序说明(这里没有给出)
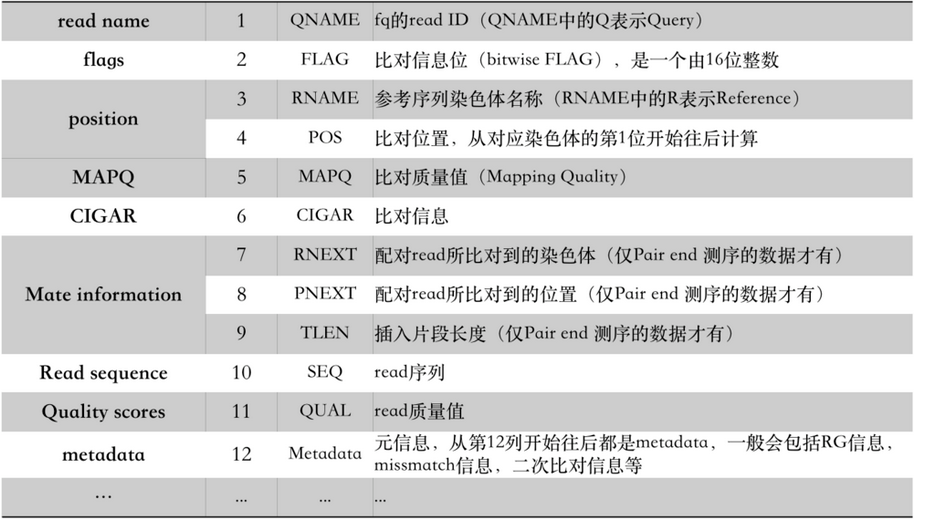
比对结果部分(alignment section)每一行表示一个片段(segment)的比对信息,包括11个必须的字段(mandatory fields)和一个可选的字段,字段之间用tag分割。必须的字段有11个,顺序固定,根据字段定义,可以为’0‘或者’*‘,这11个字段是:
1)QNAME:比对片段的(template)的编号;
2)FLAG:位标识,template mapping情况的数字表示,每一个数字代表一种比对情况,这里的值是符合情况的数字相加总和;进一步学习可查看https://broadinstitute.github.io/picard/explain-flags.html
3)RNAME:参考序列的编号,如果注释中对SQ-SN进行了定义,这里必须和其保持一致,另外对于没有mapping上的序列;
4)POS:比对上的位置,注意是从1开始计数,没有比对上,此处为0;
5)MAPQ:mappint的质量;
6)CIGAR:简要比对信息表达式(Compact Idiosyncratic Gapped Alignment Report),其以参考序列为基础,使用数字加字母表示比对结果,比如3S6M1P1I4M,前三个碱基被剪切去除了,然后6个比对上了,然后打开了一个缺口,有一个碱基插入,最后是4个比对上了,是按照顺序的;
7)RNEXT:下一个片段比对上的参考序列的编号,没有另外的片段,这里是’*‘,同一个片段,用’=‘;
8)PNEXT:下一个片段比对上的位置,如果不可用,此处为0;
9)TLEN:Template的长度,最左边得为正,最右边的为负,中间的不用定义正负,不分区段(single-segment)的比对上,或者不可用时,此处为0;
10)SEQ:序列片段的序列信息,如果不存储此类信息,此处为’*‘,注意CIGAR中M/I/S/=/X对应数字的和要等于序列长度;
11) QUAL:序列的质量信息,格式同FASTQ一样
除了上述11列外,可以有额外列:

第二列FLAG每个数值含义如下,如果符合下面多种情况,则为以下数字之和:
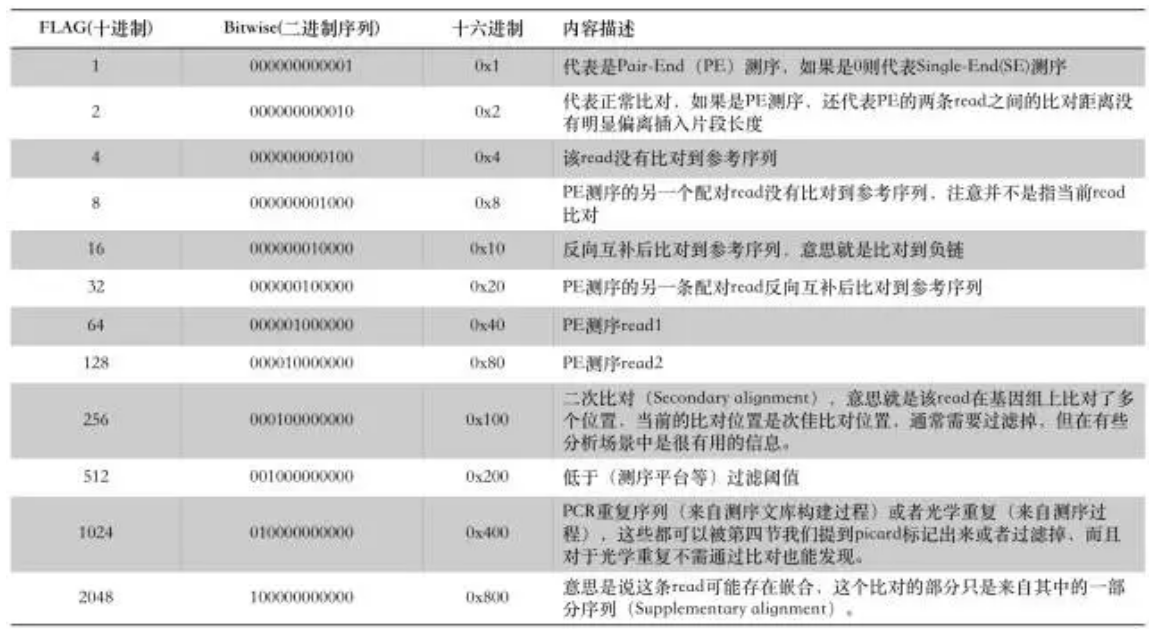
0 单端测序序列(SE)
1 (0x1) read paired read是pair中的一条(read表示本条read,mate表示pair中的另一条read)
2 (0x2) read mapped in proper pair pair一正一负完美的比对上
4 (0x4) read unmapped 这条read没有比对上
8 (0x8) mate unmapped mate没有比对上
16 (0x10) read reverse strand 这条read反向比对
32 (0x20) mate reverse strand mate反向比对
64 (0x40) first in pair 这条read是read1
128 (0x80) second in pair 这条read是read2
256 (0x100) not primary alignment 第二次比对
512 (0x200) read fails platform/vendor quality checks 比对质量不合格
1024 (0x400)read is PCR or optical duplicate read是PCR或光学副本产生
2048 (0x800)supplementary alignment 辅助比对结果
reads比对到参考序列后,bam文件中会有2048、2064这样的flag,表示supplementary alignment 。 为了理解这个概念,可能需要以下知识。
Linear Alignment
An alignment of a read to a single reference sequence that may include insertions, deletions,
skips and clipping, but may not include direction changes;(i.e. one portion of the alignment; on forward strand and another portion of alignment on reverse strand).
Chimeric Alignment
An alignment of a read that cannot be represented as a linear alignment. Typically, one of the
linear alignments in a chimeric alignment is considered the “representative” alignment,
and the others are called “supplementary” and are distinguished by the supplementary alignment flag.
1、Chimeric reads are indicative of structural variation in DNA-seq and it may indicate the
presence of chimeric genes in RNA-seq.
2、In short, chimeric reads can be split in to two or more parts, each part would be mapped
to reference(it’s not hard-clipped),the total length of the mapped part is longger than read length.
Representative alignment
A chimeric alignment that is represented as a set of linear alignments that do not have large overlaps。
Typically, one of the linear alignments in a chimeric alignment is considered the
representative alignment,and the others are called supplementary and are distinguished by the supplementary alignment.
One read can align to multiple positions, we can find one alignmnet position which sequence do not have large overlaps,
it called representative alighment, for other alignment positions,we called them supplementary alignment.
Supplementary Alignment
A chimeric reads but not a representative reads.
Primary Alignment and Secondary Alignment
A read may map ambiguously to multiple locations, e.g. due to repeats. Only one of the multiple read alignments is considered primary,
and this decision may be arbitrary. All other alignments have the secondary alignment flag.
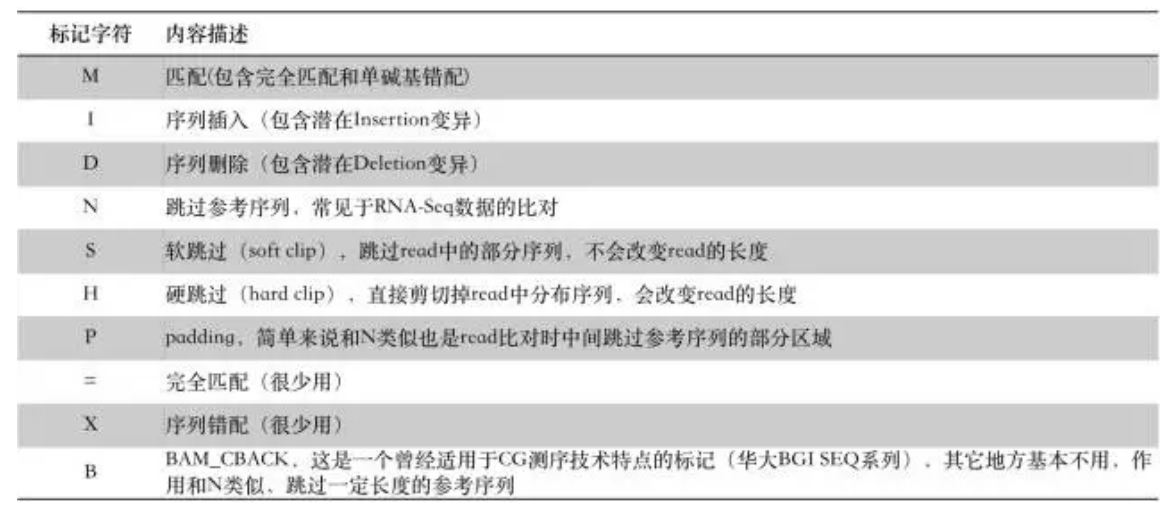
其中第六列Extended CIGAR :
M: match/mismatch
I :插入 insertion(和参考基因组相比)
D: 删除 deletion(和参考基因组相比)
N: 跳跃 skipped(和参考基因组相比)
S: 软剪切 soft clipping ,(表示unaligned,)
H: 硬剪切 hard clipping (被剪切的序列不存在于序列中)
P: 填充 padding(表示参考基因组没有,而reads里面含有位点
2)Bam (Binary Alignment/Map)
-------------------------------------------------
bam文件是Sam 文件的二进制压缩格式,保留了与sam 完成相同的内容信息。SAM/BAM 文件可以是未排序的,但是按照坐标(coodinate)排序可以线性的监控数据处理过程。samtools可以用来转化bam/sam文件,可以merg,sort aligment,可以去除duplicate,可以call snp及indels.

MAPQ:表示为mapping的质量值,等于 -10log10Probably{mapping position is wrong}, rounded to
the nearest integer, A value 255 indicates that the mapping quality is not available. 该值的计算
方法是mapping的错误率的-10log10值,之后四舍五入得到的整数,如果值为255表示mapping值是不可用
的,如果是unmapped read则MAPQ为0,一般在使用bwa mem或bwa aln(bwa 0.7.12-r1039版本)生
成的sam文件,第五列为60表示mapping率最高,一般结果是这一列的数值是从0到60,且0和60这两个数字出
现次数最多
3)对bam文件的统计
flagstat文件内容
1. in total:QC pass的reads的数量,未通过QC的reads数量为0;
2. duplicates:重复reads的数量,QC pass和failed
3. mapped:比对到参考基因组上的reads数量;
4. paired in sequencing:paired reads数据数量;
5. read1: read1的数量;
6. read2:read2的数量;
7. properly paired:正确地匹配到参考序列的reads数量;
8. 一对reads都比对到了参考序列上的数量,但是并不一定比对到同一条染色体上;
9. 一对reads中只有一条与参考序列相匹配的数量;
10. 一对reads比对到不同染色体的数量;
11. 一对reads比对到不同染色体的且比对质量值大于5的数量。
重要资料:
http://bio-bwa.sourceforge.net/bwa.shtml#4
sam/bam格式的更多相关文章
- mismatch位置(MD tag)- sam/bam格式解读进阶
这算是第二讲了,前面一讲是:Edit Distance编辑距离(NM tag)- sam/bam格式解读进阶 MD是mismatch位置的字符串的表示形式,貌似在call SNP和indel的时候会用 ...
- Edit Distance编辑距离(NM tag)- sam/bam格式解读进阶
sam格式很精炼,几乎包含了比对的所有信息,我们平常用到的信息很少,但特殊情况下,我们会用到一些较为生僻的信息,关于这些信息sam官方文档的介绍比较精简,直接看估计很难看懂. 今天要介绍的是如何通过b ...
- pysam - 多种格式基因组数据(sam/bam/vcf/bcf/cram/…)读写与处理模块(python)--转载
pysam 模块介绍!!!! http://pysam.readthedocs.io/en/latest/index.html 在开发基因组相关流程或工具时,经常需要读取.处理和创建bam.vcf.b ...
- SAM/BAM文件处理
当测序得到的fastq文件map到基因组之后,我们通常会得到一个sam或者bam为扩展名的文件.SAM的全称是sequence alignment/map format.而BAM就是SAM的二进制文件 ...
- SAMTOOLS使用 SAM BAM文件处理
[怪毛匠子 整理] samtools学习及使用范例,以及官方文档详解 #第一步:把sam文件转换成bam文件,我们得到map.bam文件 system"samtools view -bS m ...
- 文件格式——Sam&bam文件
Sam&bam文件 SAM是一种序列比对格式标准, 由sanger制定,是以TAB为分割符的文本格式.主要应用于测序序列mapping到基因组上的结果表示,当然也可以表示任意的多重比对结果.当 ...
- bwa比对软件的使用以及其结果文件(sam)格式说明
一.bwa比对软件的使用 1.对参考基因组构建索引 bwa index -a bwtsw hg19.fa # -a 参数:is[默认] or bwtsw,即bwa构建索引的两种算法,两种算法都是 ...
- pysam - 多种格式基因组数据(sam/bam/vcf/bcf/cram/…)读写与处理模块(python)
在开发基因组相关流程或工具时,经常需要读取.处理和创建bam.vcf.bcf文件.目前已经有一些主流的处理此类格式文件的工具,如samtools.picard.vcftools.bcftools,但此 ...
- 5、bam格式转为bigwig格式
1.Bam2bigwig(工具) https://www.researchgate.net/publication/301292288_Bam2bigwig_a_tool_to_convert_bam ...
随机推荐
- windows基础应用(word)
自动编号-最后选择一下编号格式 TAB 进入子节 shift+TAB 回退到父节 取消邮箱/网址链接 ctrl+z word中输入不认识生僻字:输入偏旁部首王,选中插入中寻找 输入英文时,更改大小写/ ...
- RF安装
参考: http://www.cnblogs.com/zlj1992/p/6357373.html https://github.com/robotframework/RIDE/wiki/Instal ...
- Linux下不同颜色文件的类型
蓝色表示目录: 绿色表示可执行文件: 红色表示压缩文件: 浅蓝色表示链接文件:主要是使用ln命令建立的文件 灰色表示其它文件: 红色闪烁表示链接的文件有问题了: 黄色是设备文件,包括block, ch ...
- javascript面向对象的程序设计之Object.getOwnPropertyDescriptor()
Object.getOwnPropertyDescriptor()用于获取给定属性的描述信息,这个描述信息是一个对象. 如果是访问器属性,则这个对象的属性有configurable,enumerabl ...
- Scrapy-下载中间件
下载中间件 下载器中间件是介于Scrapy的request/response处理的钩子框架. 是用于全局修改Scrapy request和response的一个轻量.底层的系统 编写您自己的下载器中间 ...
- 【BZOJ】1085 [SCOI2005]骑士精神(IDA*)
题目 传送门:QWQ 分析 我好菜啊. 一波IDA*水过去了. 代码 #include <bits/stdc++.h> using namespace std; ; char s[maxn ...
- R语言中的遗传算法详细解析
前言 人类总是在生活中摸索规律,把规律总结为经验,再把经验传给后人,让后人发现更多的规规律,每一次知识的传递都是一次进化的过程,最终会形成了人类的智慧.自然界规律,让人类适者生存地活了下来,聪明的科学 ...
- django中的 form 表单操作
form组件 1. 能做什么事? 1. 能生成HTML代码 input框 2. 可以校验数据 3. 保留输入的数据 4. 有错误的提示 1. 定义 from django ...
- List集合的clear方法
一 . list.clear()底层源码实现 在使用list 结合的时候习惯了 list=null :在创建这样的方式,但是发现使用list的clear 方法很不错,尤其是有大量循环的时候 1.lis ...
- tornado-模板继承extend,函数和类的导入
大 import tornado.ioloop import tornado.web import tornado.httpserver # 非阻塞 import tornado.options # ...