ONCOCNV软件思路分析之tumor处理
前期处理
- perl脚本统计RC(RC(read counts))
- 读入control baseline 和 sigma(最后baseline 预测的mad值)
- 将gc < 0.28或gc > 0.68,sigma乘上1.5,后来又乘以6,对于小于0.01或者大于0.99分位数,sigma取0.01和0.99分位点的sigma
- 将sigma转化为权重,SigmaForWeights = 1/sigma2/max(1/sigmaforWeithts2)
- 根据mu值设置一些outlier的amplicon,threshold为-2和2
- 如果1/3 amplicon都是NA,该样品被抛弃
文库大小校正,(中位数校正),GC含量校正,长度校正,ICA校正
- 文库大小校正,(中位数校正),GC含量校正,长度校正,多了一步中位数校正,将NRC除以总的NRC值的中位数,作用是一开始对于0拷贝数进行校正
- ICA标准化:取control样品中的ICA1,ICA2,ICA3使用rlm建立线性模型求残差作为标准化 后的值,对于值为0的logNRC,取最小logNRC
segmentByCBS
- 输入标准化后的各个amplicon的染色体,起始位置,logNRC和权重(SigmaForWeights),使用PSCBS中segmentByCBS(内部调用DNAcopy包)函数进行片段划分。划分时,sigma越大权重越小,来避免断点判定在sigma较大的amplicon。输出是每个segment对应的拷贝数
输出如下,如果该染色体中存在NA的数据,会保留一行NA的值
sampleName chromosome start end nbrOfLoci mean
1 <NA> 1 2488068 244006378 1363 0.0934
2 <NA> NA NA NA NA NA
3 <NA> 2 5833035 234681120 1008 0.0732
4 <NA> NA NA NA NA NA
5 <NA> 3 3192502 195622096 939 0.0718
6 <NA> NA NA NA NA NA
7 <NA> 4 1800963 153332857 423 0.0313
8 <NA> NA NA NA NA NA
9 <NA> 5 223515 180058652 574 -0.3460
10 <NA> NA NA NA NA NA
11 <NA> 6 393195 167275553 1378 0.0588
12 <NA> NA NA NA NA NA
13 <NA> 7 2946229 152373106 945 0.0507
14 <NA> NA NA NA NA NA
15 <NA> 8 30915961 145742416 852 0.0803
16 <NA> NA NA NA NA NA
17 <NA> 9 5021975 5072501 21 -0.1446
18 <NA> 9 5072501 134100903 474 -0.3635
19 <NA> 9 134100903 139438447 156 0.0474
predictCluster,,确定zero(copy number 0对应的logNRC)和copy number。注:这里求出的copy number并不是生物意义的copynumber 而是是否出现拷贝数异常,0为未出现拷贝数异常
- 将每个segment内的amplicon乘以权重求中位值得到权重后得到的weighted.median segmean,如果无法求出weighted.median,取wighted.mean作为segmean
- 原理:对应着相同copy数的segmean应该聚类在一起
- 因为直接取segmean进行聚类可能会聚类错误,因此对数据添加高斯噪音:
第一步,求出需要添加的sdError:取出在-2和2之间的amplicon(并且排除x和y染色体),将amplicon的logNRC - segmean得到errors,对每一段segment的errors求方差,得到该段sdNoise,再将sdNoisd比上该段amplicon长度开根得到sdError,公式为:sdError = sd(logNRC - segmean)/sqrt(n),可以理解为sdError = ((logNRC - segmean) - mean(logNRC - segmean))^2/sqrt( n(segment)*n(in each segment) )
第二步,添加噪音,噪音的均值为0,方差为sdError,每个ampliocn 的值a = segmean + rnorm(n(in each segment),0,sdError) - 使用mcluser聚类
如果聚类结果大于1类,将该所有类别的方差取出,将异常簇给值NA,异常cluster判断方法:每个cluster都有一个对应的标准差(sigma),标准差大于中位cluster sigma加上7*mad(sigma) - 计算每个cluster的density
计算方法:计算该cluster(高斯模型)的density,等于每个cluster(全部高斯模型)的均值在这个cluster下的density的总和再乘以这个cluster的density比重。而每个cluster的density = dnorm(x,mean of cluster,var of cluster) * proportion of cluster - 将最大density的cluster作为zero copy number
- 将最近的一个cluster(如果mean值差<0.04)两个cluster乘比重后的均值赋给该cluster(zero),该cluster比重变为这两个cluster相加

- 检测预测时处于边缘的值
如果同一个amplicon a 对应的copy number mad值为0.5,那么a所对应的amplicon copy number 为1,如果同一个amplicon a 对应的copy number mad值为-0.5,那么a所对应的amplicon copy number 为-1
如果常染色体只有1个cluster,那么分析x,y染色体,如果segmean < log(0.75),该amplicon copy number 给-1,如果segmean > log(1.25),amplicon copy number给1 - 求出maxloss 和 maingain
maxloss:
copy number是-1的amplicon的segmean,
copy number 是0 的amcplion segmean -(copy number是0的amplicon segmean - mean(copy number 是 -1的 amplicon segmean ))/2
在上述中取最大值
mingain:
copy number是1的amplicon的segmean,
copy number 是0 的amcplion segmean +(mean(copy number 是 1的 amplicon segmean )-copy number是0的amplicon segmean)/2
在上述中取最小值 - 将x和y染色体segmean > mingain ,copy number赋予1,将x和y染色体segmean < maxloss,copy number 赋予-1
- 对predictCluster进行八次,取最小的zero值的一次,然后取出zero和预测的copy number
计算生物意义的copy number
- ratio = logNRC - zero,减去zero值(copy number 0 的cluster的均值)
- 如果存在正常细胞污染的比例,那么减去这部分的影响: ratio = log((exp(ratio)-normalContamination)/(1-normalContamination))
- 求出copy number 0的amplicon求mad值,作为全部的标准差(sample sigma),所有amplicon的control sigma * sample sigma作为校正后的方差
- 将每个segment内的amplicon ratio乘以权重求中位值得到权重后得到的weighted.median segmean,如果无法求出weighted.median,取wighted.mean作为segmean
- copy number计算方法
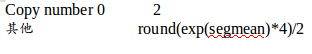
按照segment进行fixed variance test和t test
- fixed variance test
ratio 应服从N(0,sigma),对segment内的amplicon ratio 进行转换,(logNRC-0)/sigma应该服从N(0,1),根据中心极限定理,mean((logNRC-0)/sigma)服从N(0,1/sqrt(n)),计算该segment的mean((logNRC-0)/sigma)在N(0,1/sqrt(n))双尾的p值 - t test
(logNRC-0)/sigma应该服从N(0,1),对每个amplicon进行t test
如果fixed variance test > 0.01 或者t test > 0.01,copy number赋予2
CNV:copy >= 3 : Gain,copy = 2.5:PotentialGain,copy < 1:Loss,copy = 1.5:PotentialLoss
检测segment的outlier
(ratio- segmean)/sigma在N(0,1)双尾的p值
ratio/sigma在N(0,1)双尾的p值
取两者之中较大的p值作为该amplicon的outlier p值
所有的amplicon outlier值采用fdr方法计算q值
对于outlier q值<0.01的点加上注释,outlier p值,outlier copy number采用round(exp(values[tt])*4)/2
按照基因进行t test
- 对每个基因使用t检验,理论上 ratio/sample sigma/control sigma,服从均值为0的分布,如果p值<0.01,对每个基因copynumber进行估计(perGeneEvaluation)
- 如果基因计算的结果不等于segment的结果,并且断点不在基因内,ratio - segmean / (control sigma * sample sigma)应服从1/sqrt(n)正态分布,作t检验,如果t检验显著,predLarge Corrected给perGeneEvaluation
- 如果断点在基因内,以第一个segmean(fragratio)作为参考,如果出现segmean不一样,将下一个amplicon segmean值改为参考segmean
- 对每个基因内的ratio - segmean(fragratio)/(control sigma * sample sigma),作t检验,取出最大的p值作为基因内所有amplicon的pvalRatioCorrected
- 但如果基因内存在copy number = 2 的amplicon,判断是否存在round(exp(fragratio)*4)/2 = 2,如果有从这些amplicon中取出p值最大的p值作为基因内所有amplicon的pvalRatioCorrected
- 如果该p值>0.05,说明全部点都被该fragratio解释了
- 如果p值<0.05,说明不能全部解释,对每个fragratios相同的amplicon取出,将它们ratio/(control sigma * sample sigma)作t检验,如果最小值>0.01,那么pergeneEvaluation=2
参考资料
ONCOCNV文献:https://academic.oup.com/bioinformatics/article/30/24/3443/2422154
ONCOCNV代码:http://boevalab.com/ONCOCNV/
ONCOCNV软件思路分析之tumor处理的更多相关文章
- ONCOCNV软件思路分析之control处理
进行数据初步处理(perl) 统计amplicon的RC(read counts),并且相互overlap大于75%的amplicon合并起来 统计每个amplicon的GC含量,均值, 性别识别并校 ...
- 福州大学软件工程1816 | W班 第10次作业[个人作业——软件产品案例分析]
作业链接 个人作业--软件产品案例分析 评分细则 本次个人项目分数由两部分组成(课堂得分(老师/助教占比60%,学生占比40%)满分40分+博客分满分60分) 课堂得分和博客得分表 评分统计图 千帆竞 ...
- 书评第003篇:《0day安全:软件漏洞分析技术(第2版)》
本书基本信息 丛书名:安全技术大系 作者:王清(主编),张东辉.周浩.王继刚.赵双(编著) 出版社:电子工业出版社 出版时间:2011-6-1 ISBN:9787121133961 版次:1 页数:7 ...
- laravel中间件的创建思路分析
网上有很多解析laravel中间件的实现原理,但是不知道有没有读者在读的时候不明白,作者是怎么想到要用array_reduce函数的? 本文从自己的角度出发,模拟了如果我是作者,我是怎么实现这个中间件 ...
- 软件案例分析——VS、VS Code
软件案例分析--VS和VS Code 第一部分 调研,测评 一.使用10–30分钟这个软件的基本功能(请上传使用软件的照片) VS code Visual Studio 二.主要功能和目标用户有何不同 ...
- 软件产品案例分析--K米
软件产品案例分析--K米 第一部分 调研,评测 评测 个人第一次上手体验 使用的第一款点歌软件,以为就是个遥控而已,使用后发现功能还挺多,能点挺久.觉得很方便,不用挤成一堆点歌了.K米的脸蛋(UI)好 ...
- 团队项目2.0软件改进分析MathAPP
软件改进分析 在此基础上,进行软件的改进. 首先,我们把这个软件理解成一个投入市场的.帮助小朋友进行算术运算练习的APP. 从质量保证的角度,有哪些需要改进的BUG? 从用户的角度(把自己当成小学生或 ...
- [团队项目2.0]软件改进分析MathAPP
软件改进分析 在此基础上,进行软件的改进. 首先,我们把这个软件理解成一个投入市场的.帮助小朋友进行算术运算练习的APP. 从质量保证的角度,有哪些需要改进的BUG? 从用户的角度(把自己当成小学生或 ...
- 第五周课后作业——热门软件创新分析+附加题1&附加题3
鉴于我们寝室都热衷于手游,所以本次热门软件创新分析我就来分析一下几款热门的抽卡型手游. 阴阳师(后文简称YYS)——剧情画风唯美,配音引人入胜 作为网易公司研发的一款3D日式和风回合制游戏,YYS ...
随机推荐
- 洛谷 [P1220] 关路灯
本题是一道区间DP,很容易设计出状态, dp[i][j]代表关掉i到j的路灯所耗的电量,但是对于新到一个路灯来说,可以是原来直接来的,也可以是掉头来的,于是还需要添加一维 0代表在区间的左端,1代表在 ...
- BZOJ 4034: [HAOI2015]树上操作 [欧拉序列 线段树]
题意: 操作 1 :把某个节点 x 的点权增加 a . 操作 2 :把某个节点 x 为根的子树中所有点的点权都增加 a . 操作 3 :询问某个节点 x 到根的路径中所有点的点权和. 显然树链剖分可做 ...
- 事务与隔离级别------《Designing Data-Intensive Applications》读书笔记10
和数据库打交道的程序员绕不开的话题就是:事务,作为一个简化访问数据库的应用程序的编程模型.通过使用事务,应用程序可以忽略某些潜在的错误场景和并发问题,由数据库负责处理它们.而并非每个应用程序都需要事务 ...
- 微信小程序页面跳转的问题(app.json中设置tarBar后wx.redirectTo和wx.navigateTo均不能实现跳转到指定的页面)
1.设置的tabBar代码片段: "tabBar": { "list": [ { "pagePath": "pages/homep ...
- Azure Automation (6) 执行Azure SQL Job
<Windows Azure Platform 系列文章目录> 因为China Azure SQL Database目前还没有SQL Job的功能,如果要异步执行SQL 存储过程,可以使用 ...
- 小甲鱼OD学习第3讲
这次我们的任务是破解这个过期的软件,效果如图所示 我们通过阅读代码,知道这个程序的执行流程如图中注释所示 观看下图注释所示 这是失败的提示代码 这是成功的提示代码 最后我们可以得出结论,成功破解软件的 ...
- 【模板小程序】循环方阵构造(仿《剑指offer》循环矩阵打印)
/* 本程序说明: 输入:方阵大小n,输出:n*n的旋转方阵 举例: 当n=2时,输出: 1 2 4 3 当n=4时,输出: 1 2 3 4 12 13 14 5 11 16 15 6 10 9 8 ...
- CentOS 7 使用iptables防火墙
# 停止firewalld服务 systemctl stop firewalld systemctl mask firewalld # 安装iptables-services yum install ...
- iOS视频直播
视频直播技术点 视频直播,可以分为 采集,前处理,编码,传输, 服务器处理,解码,渲染 采集: iOS系统因为软硬件种类不多, 硬件适配性比较好, 所以比较简单. 而Android端市面上机型众多, ...
- jdk 1.8 开发环境配置
计算机->右键->属性->高级系统设置->环境变量->系统变量 新建系统变量:JAVA_HOME,变量值为:C:\Program Files (x86)\Java\jdk ...