一次rna-seq的过程-知乎live转
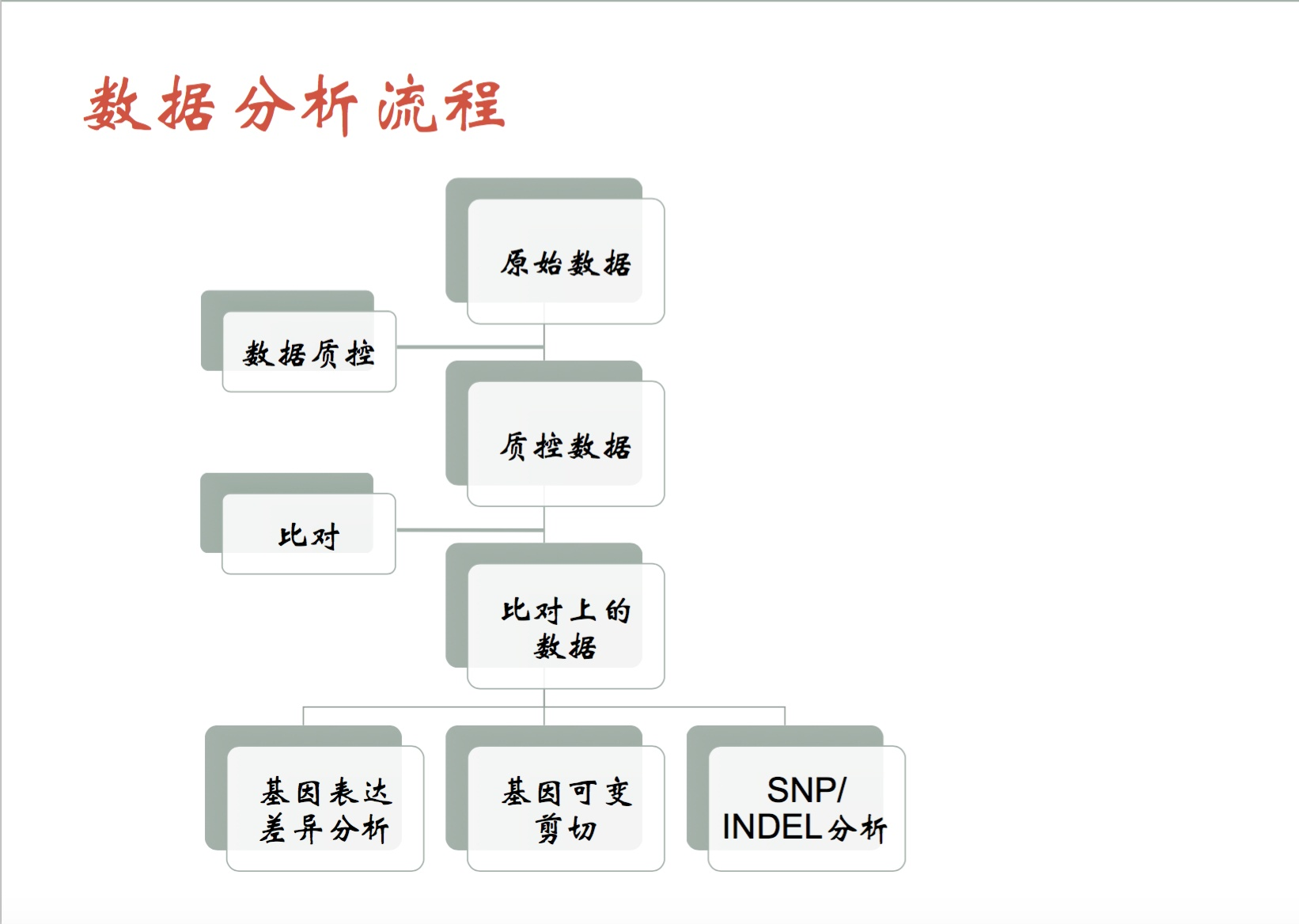
数据分析流程
来自知乎孟浩巍的“快速入门生物信息学的”Live,超棒的~
1.数据质控
首先是质控部分,使用fastqc进行对结果分析。
对于Illumia二代测序的结果质控包括两个方面,去掉测序质量不好的序列,即Quality Control;二是需要去掉连在玻璃上的短的接头,cut adaptor。

-t 8表示调用8个核心去运算。
之后,对每一个序列文件都生成一个zip和一个html文件。
例如:
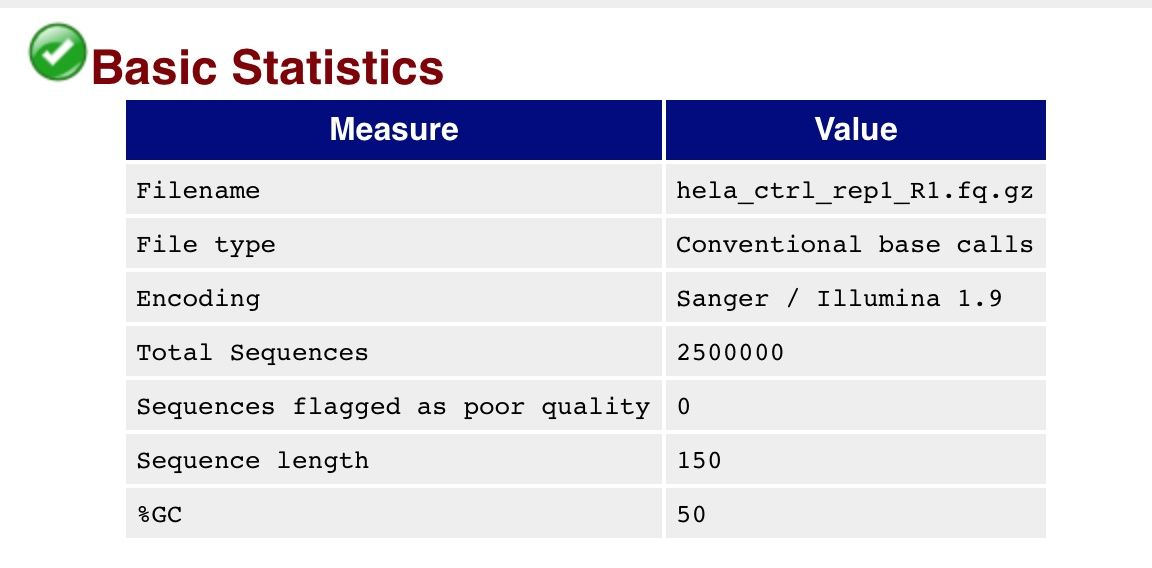
那么这2500000肯定是不同的基因,只不过这个机器的测序长度是150,所以所有的基因长度都是相同的。
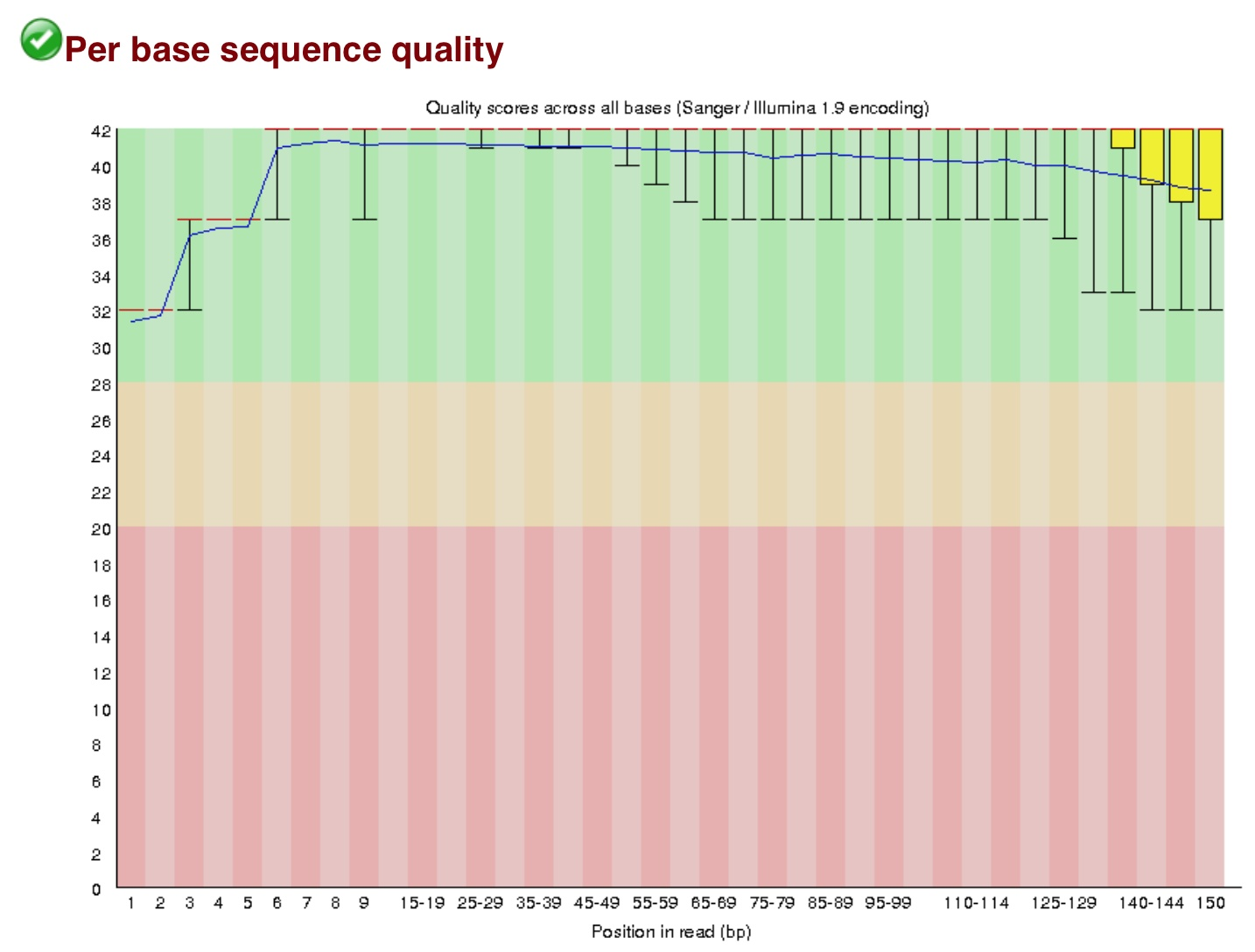
对250万reads,每个位点的Q做一个箱线图,要求箱线值最低点高于20%,否则需要将那部分切除。
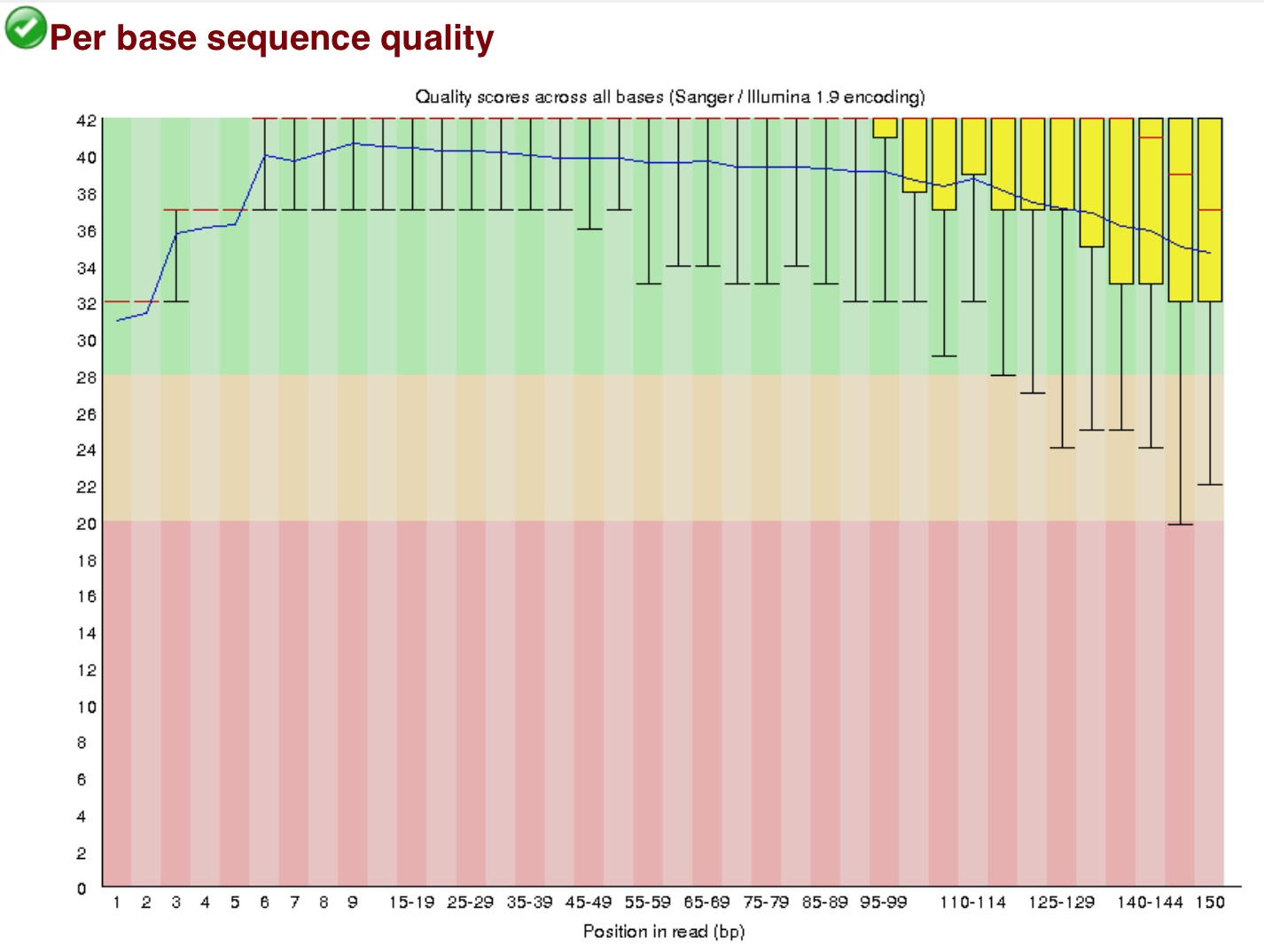
在145左右的的序列Q值较低测序不稳,所以这样的序列145之后的全不要了。

这个是GC含量图,通常A和T相同,C和G相同,但是前10bp不稳定,需要切除。
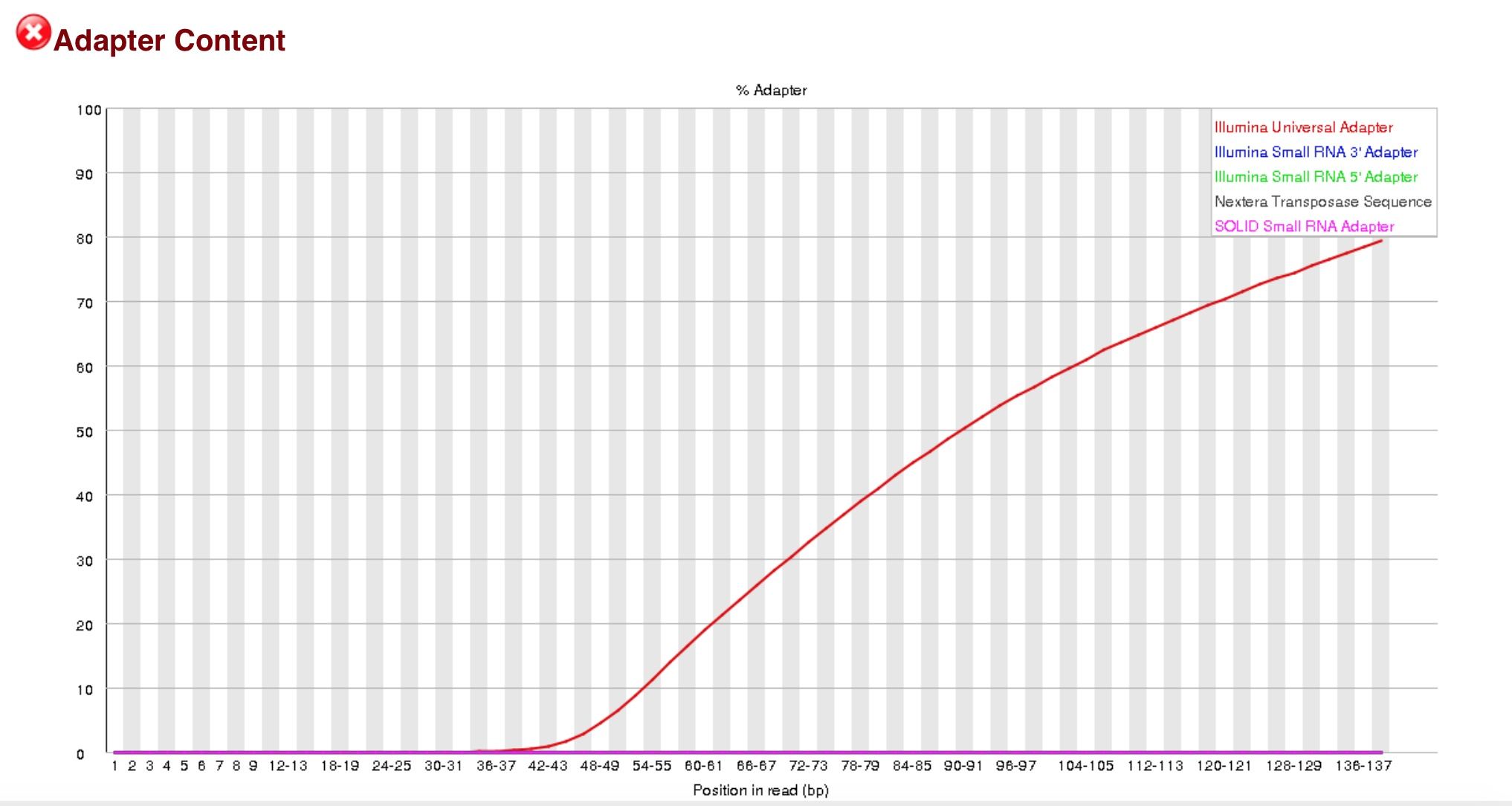
表示测序过程中测到的段序列的含量,横轴是1-150bp,纵轴是百分比。由于某种原因导致测的绝大多数都是测的adaptor。
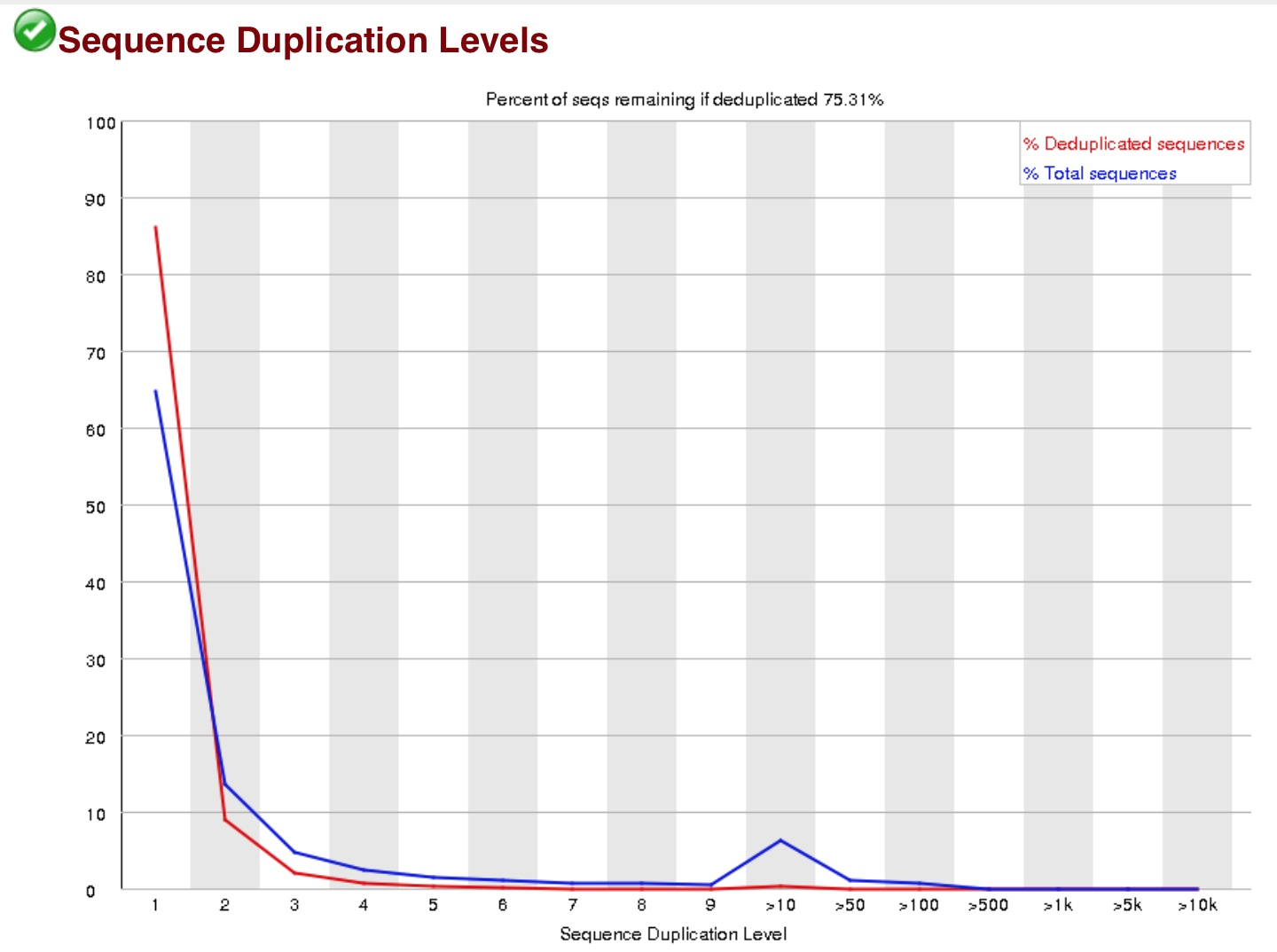
这个主要是衡量建库水平,建库中通常有6-8轮的PCR,但有时会出现过P的现象,当duplication过高的情况下,需要去dup。但是在RNA-seq里通常是不去dup的。
②接下来,使用fastx_trimmer去头去尾。

zcat $fastq_1 | fastx_trimmer -f 11 -l 140 -z -o $out_fastq_1 &
zcat解压缩,$fastq_1是输入的第一个文件,这个文件解压缩之后的结果给fastx_trimmer这个命令,
这个命令的参数-f是指first即保留的第一个bp(这里前10bp剪切掉了);last即保留的最后一个bp(保留到第140bp),-z是压缩命令,-o是输出到这个文件里。
//其中$:在bash里表示当前是普通用户;是变量引用操作符。a=10; echo $a会输出10。
③使用cutadaptor去掉两端的adaptor。
trimmer之后有一个去adaptor的过程,使用cutadaptor的软件,

nohup cutadapt --times 1 -e 0.1 -0 3
--quality-cutoff 6 -m 50 -a AGATCGGAAGAGC
-A AGATCGGAAGAGC -o $out_fastq_1
-p $out_fastq_2 $fastq_1 $fastq_2 > $log_file 2>$1 &
//times 1一条序列只去一次Adaptor;-e 0.1在匹配时可以有10%的错误率;-O 3 adaptor序列必须和测序序列有3个碱基以上的overlap才可以;常用6;-m 50如果处理之后低于50的话就扔掉序列,短序列测序质量可能不是很好;-a和-A是Illumina常用的通用引物,之所以输入两个,是因为我是一个双端测序的结果,需要对两个文件内容进行分别去除,-a对应Reads1,-A对应reads2,$fasrq_1和_2是上一步的输出;>最后是写入log文件
//其中nohup:不挂断地运行命令。
//2>$1:$1是传递给shell脚本的第一个参数;(转自:https://www.cnblogs.com/kaituorensheng/p/4002697.html)
$# 是传给脚本的参数个数
$0 是脚本本身的名字
$1 是传递给该shell脚本的第一个参数
$2 是传递给该shell脚本的第二个参数
$@ 是传给脚本的所有参数的列表
$* 是以一个单字符串显示所有向脚本传递的参数,与位置变量不同,参数可超过9个
$$ 是脚本运行的当前进程ID号
$? 是显示最后命令的退出状态,0表示没有错误,其他表示有错误
例子:
##dels.sh
echo "number:$#"
echo "scname:$0"
echo "first :$1"
echo "second:$2"
echo "argume:$@"
echo "show parm list:$*"
echo "show process id:$$"
echo "show precomm stat: $?"
[@jihite]$ sh del.sh
number:
scname:del.sh
first:
second:
argume:
show parm list:
show process id:
show precomm stat:
//Linux中文件重定向符> <
>覆盖文件;>>追加内容;如果重定向输出的文件不存在,则会新建文件。
<从文件中读入;
2>将命令执行过程中出现的错误信息(选项或者参数错误) 保存到指定的文件中,而不是直接显示到显示器;2是指错误文件的编号(标准的输入输出中省略了1 0编号)
每个进程都和三个系统文件相关联:标准输入stdin,标准输出stdout和标准错误stderr,三个系统文件描述分别为0,1和2.所以这个意思是将标准错误也输出到标准输出中。
//cat命令
三大功能:1.一次显示整个文件,cat filename;2.从键盘创建一个文件,cat > filename;3.将几个文件合并为一个文件,cat file1 file2 > file。
// ./是什么意思?
表示当前目录,./aaa表示执行在当前目录下的aaa。./后跟脚本文件,用来执行脚本。
④将RNA序列比对到rRNA上,保留不能比对到rRNA的结果。

nohup bowtie2 -x $rRNA_index - $fastq_1 - $fastq_2 -S $sam_out -p --un -c
onc-gz $fastq_unmap > $log >& &
一般通过map到rRNA中的比例来衡量建库的质量。一般的要求rRNA的比例不超过10%。
rRNA就是核糖体RNA, 是3类RNA中相对分子质量最大的,是在mRNA的指导下将氨基酸合称为肽链,rRNA占RNA总量的82%。单独存在时不执行功能,多与蛋白质结合在一起形成核糖体。
//核糖体:是细胞中的一种细胞器,主要由RNA和蛋白质构成,其唯一功能是按照mRNA的指令将氨基酸合成蛋白质多肽链,所以核糖体是细胞内蛋白质合成的分子机器。
//-x对应rRNA的索引序列;-1,-2是刚输出的reads1和reads2;-S是比对结果的输出文件;-p 4使用四个核心去运算(p就是processor吧!);一长串-gz,输出比对不上的结果;
⑤随后需要使用tophat2将过滤掉的reads比对到ref基因组上
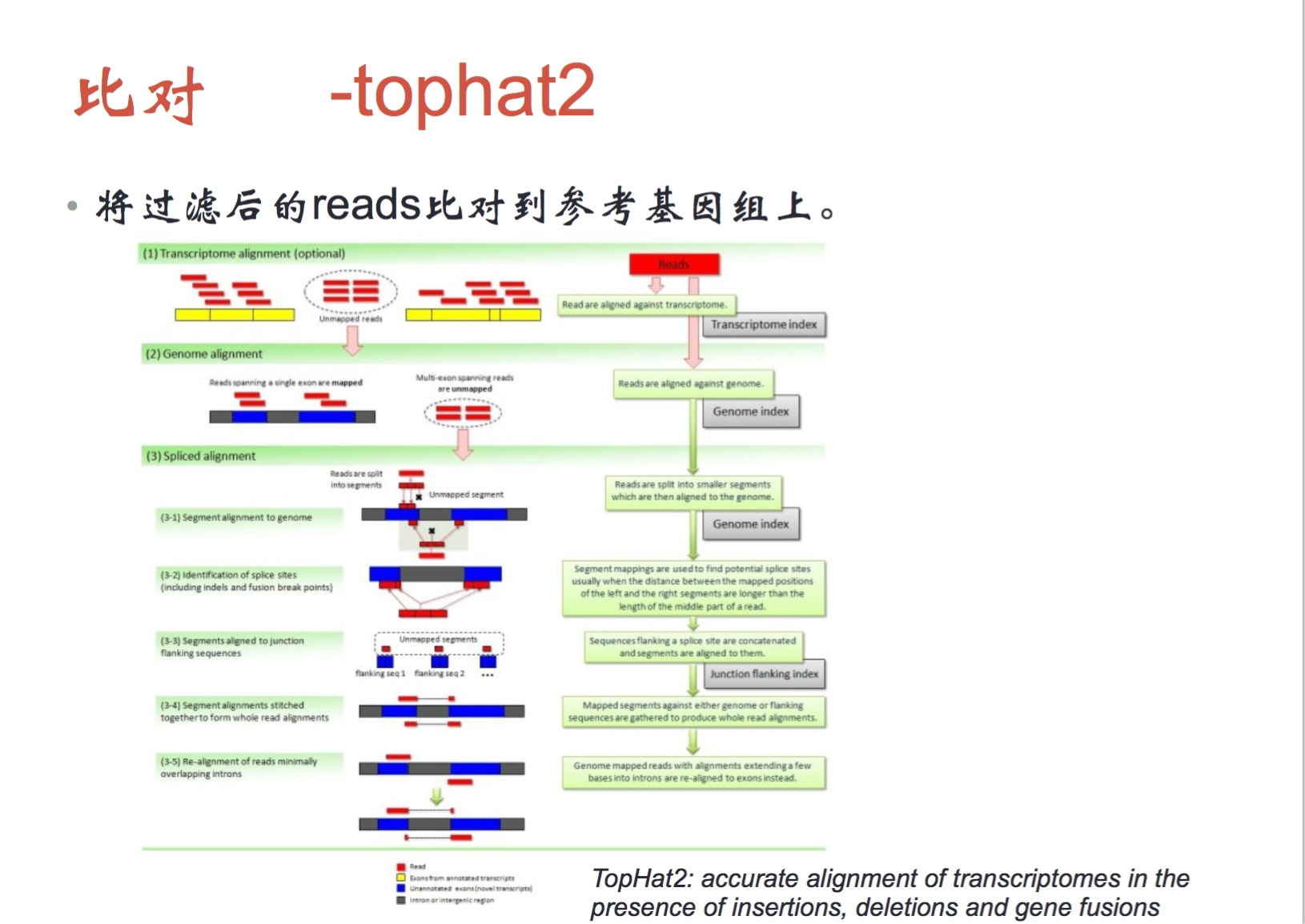
如果mRNA直接比对到人的DNA上,可能会出现问题,有可能跨越了一个内含子,tophat2考虑了这个问题,它将reads根据注释文件分开成短序列,重新比对;

nohup tophat2 -p -o $output_dir $h19_index $fastq_1 $fastq_2 > $log >& &
//$output_dir是一个文件夹,输出结果到这个文件夹中,h19是人的基因组版本,包括h19和h38,h19包含信息丰富。
//$fastq_1 $fastq_2是质控完没有比对到rRNA上的序列。
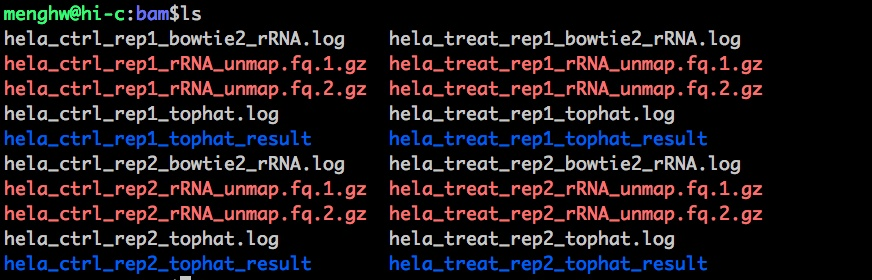
这个是最终的结果,有map到rRNA的,有没有map到rRNA的;蓝色的文件夹就是tophat2的运行结果。

那么将蓝色文件夹展开:
.bam是最终的比对结果;.txt是比对中的总结情况;.info没有直接map到连续的基因组上,需要切一些reads,加工reads的文件保存在info里;unmapped.bam是一层层都没有比对到的,可能是基因组上未注释过的、测序问题。
下面是bam文件的讲解: 头部和比对信息,bam是压缩格式;
⑥得到bam文件后,对基因表达量进行评估

nohup cufflinks -o $cufflink_dir -p -G $hg19_gtf $bam_file > $log >& &
2020-4-30更新——————————
https://www.jianshu.com/p/9c99e09630da
到底什么是bam文件,和sam有什么不同,
bam是sam的二进制,SAM是一种序列比对格式标准,SAM分为两部分,注释信息(header section)和比对结果部分(alignment section)。
具体的我都没往下看,太麻烦了。
//-o输出到文件夹里,-G是需要用的转录组的参考文件,需要输入bam文件
这个是cufflink的输出文件。fpkm是衡量基因表达量的数值,一个基因有不同的内含子和外显子,不同的外显子之间可以形成不同的转录本,每一个转录本可以翻译成不同的蛋白,这些蛋白互相之间就是isoforms(亚型),对于不同的转录本来说基因有一个表达量,这就是基因的fpkm和isoform的fpkm。
基因表达量详解:
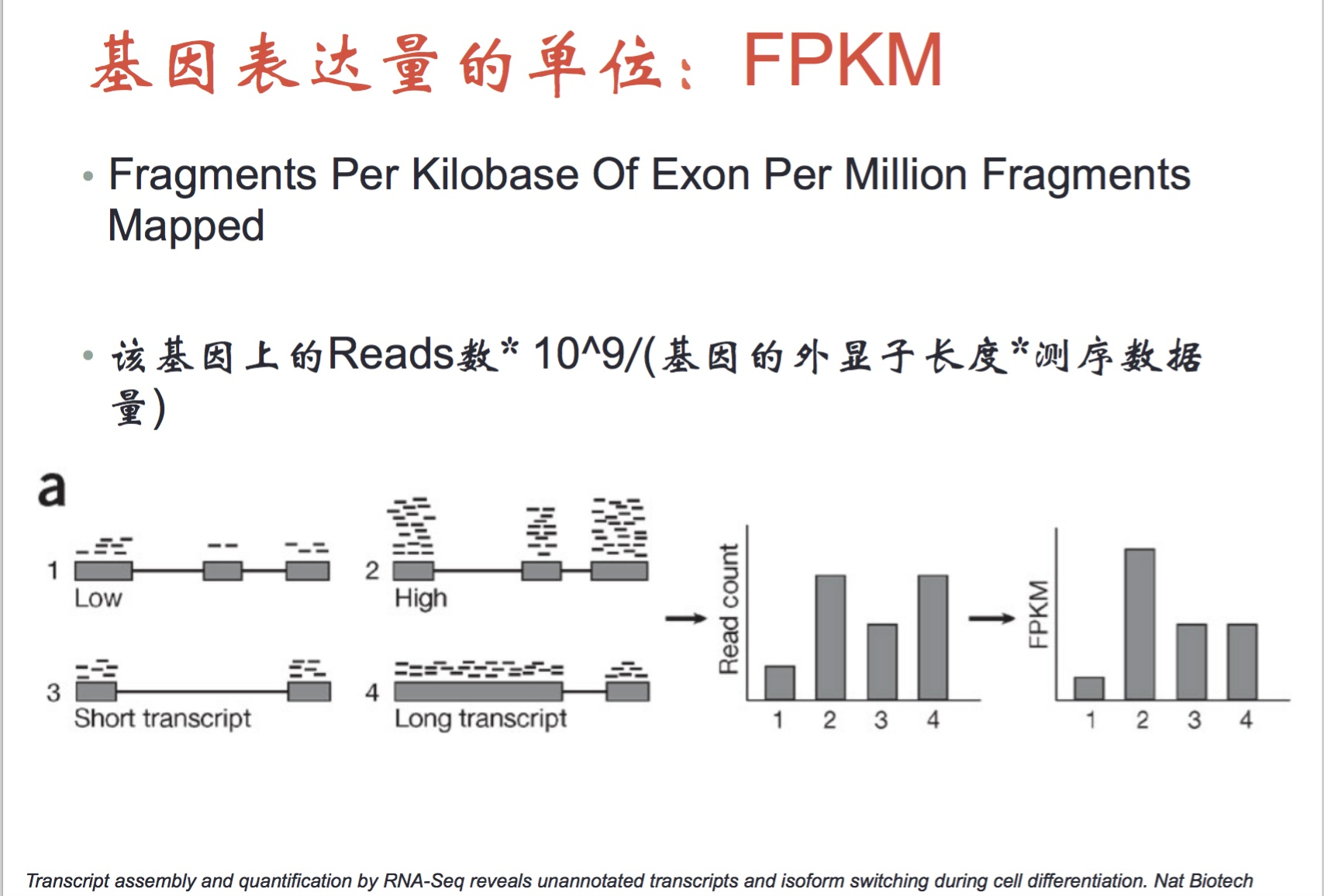
一对pair可以确定一个fragment,每测1Mreads,平均1kb上就能回帖多少reads,这个reads的数量就是对应的fpkm数值。
【每一个外显子上比对了多少reads/整个的外显子整体长度(以kb为单位)/总的测序量(以M为单位)】
校正了基因的长短。
⑦计算基因表达差异,使用cuffdiff


nohup cuffdiff -o $out_dir -p --labels $label --min -reps-for-js-test $hg19_
gtf $ctrl_bam $treat_bam > $log >& &
--lables是文件的输入次序,如上label=hela.ctrl,hela_treat;--min 每个treat里有几个repeat,你看上边ctrl_bam是两个,要和treat_bam数量一致且>=2。
这是运行结果,对gtf中有的基因都进行了检测,
//其中cds是:蛋白质编码区。
cuffdiff做了一个比较特殊的T-test。
⑧在R中处理基因表达差异


locus是基因的范围;sample1一般是ctrl组,sample2一般是treatment组;status中notest说至少有一个fpkm没算出来,OK是指两个reads的都较可信;q_value是p_value的矫正值,<0.05一般认为显著。
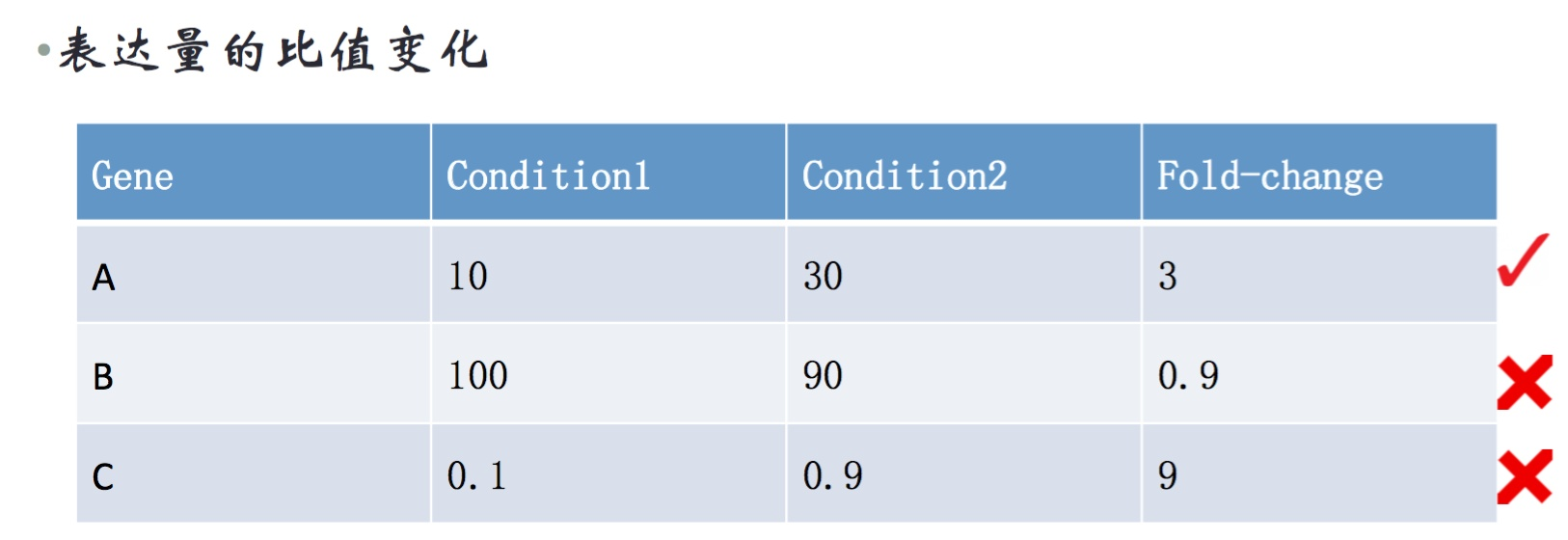
fold-change是样本质检表达量的差异倍数。一般要求fold-change>2,并且condition1和2要同时>1,或者至少有一个fpkm>1。
严格的标准是:1, log2(treat/ctrl)的绝对值大于1;2. FPKM要都大于1。
一次rna-seq的过程-知乎live转的更多相关文章
- RNA -seq
RNA -seq RNA-seq目的.用处::可以帮助我们了解,各种比较条件下,所有基因的表达情况的差异. 比如:正常组织和肿瘤组织的之间的差异:检测药物治疗前后,基因表达的差异:检测发育过程中,不同 ...
- RNA seq 两种计算基因表达量方法
两种RNA seq的基因表达量计算方法: 1. RPKM:http://www.plob.org/2011/10/24/294.html 2. RSEM:这个是TCGAdata中使用的.RSEM据说比 ...
- 生物信息学——RNA的剪切过程
生物信息学——RNA的剪切过程 外显子(exon expressed region)是真核生物基因的一部分,它在剪接(Splicing)后仍会被保存下来,并可在蛋白质生物合成过程中被表达为蛋白质. ...
- RNA测序相对基因表达芯片有什么优势?
RNA测序相对基因表达芯片有什么优势? RNA-Seq和基因表达芯片相比,哪种方法更有优势?关键看适用不适用.那么RNA-Seq适用哪些研究方向?是否您的研究?来跟随本文了解一下RNA测序相对基因表达 ...
- linux0.12 编译过程
感谢这篇文章的作者: http://www.cnblogs.com/strugglesometimes/p/4231359.html 编译是个很蛋疼的事情,本想把linux0.12在bochs上 ...
- 为你揭秘知乎是如何搞AI的——窥大厂 | 数智方法论第1期
文章发布于公号[数智物语] (ID:decision_engine),关注公号不错过每一篇干货. 数智物语(公众号ID:decision_engine)出品 策划.编写:卷毛雅各布 「我们相信,在垃圾 ...
- xgene:WGS,突变与癌,RNA-seq,WES
人类全基因组测序06 SNP(single nucleotide polymorphism):有了10倍以上的覆盖深度以后,来确认SNP信息,就相当可靠了. 一个普通黄种人的基因组,与hg19这个参 ...
- 让DB2跑得更快——DB2内部解析与性能优化
让DB2跑得更快——DB2内部解析与性能优化 (DB2数据库领域的精彩强音,DB2技巧精髓的热心分享,资深数据库专家牛新庄.干毅民.成孜论.唐志刚联袂推荐!) 洪烨著 2013年10月出版 定价:7 ...
- php大力力 [025节] 来不及学习和分类的,大力力认为有价值的一些技术文章合集(大力力二叔公)(2015-08-27)
php大力力 [025节] 来不及学习和分类的,大力力认为有价值的一些技术文章合集(大力力二叔公)(2015-08-27) 比较好的模版 免费模板网,提供大量DIV+CSS布局网页模板下载及后台管理 ...
随机推荐
- c# 文件操作 txt、xml、ini
1. txt文件 /// <summary> /// 读文本文件信息 /// </summary> /// <param name="FilePath" ...
- HDU 1075 What Are You Talking About (Trie)
What Are You Talking About Time Limit: 10000/5000 MS (Java/Others) Memory Limit: 102400/204800 K ...
- cocos2d-x 输入框CCEditBox的使用
特别说明: 这个版本的CCEditBox,设计有缺陷,背景图片的位置与输入区域的位置不同步,需要自己修改原来的代码,自己加上输入区域的坐标偏移量. void CCEditBox::setPositio ...
- 小程序WXML 使用小结
数据绑定 <view> {{message}} </view> // page.js Page({ data: { message: 'Hello MINA!' } }) 组件 ...
- SpringMVC返回Json,自定义Json中Date类型格式
http://www.cnblogs.com/jsczljh/p/3654636.html —————————————————————————————————————————————————————— ...
- COCOS2D-HTML5 开发之二】cocos2d-html5项目定义成员,局部变量,函数笔记随笔
本站文章均为李华明Himi原创,转载务必在明显处注明:(作者新浪微博:@李华明Himi) 转载自[黑米GameDev街区] 原文链接: http://www.himigame.com/cocos2d- ...
- jQuery-对Select的操作集合[终结篇]
jQuery获取Select选择的Text和Value: 请选择 C# Javascript jQuery C++ Java VB 选择一项试试看语法解释:1. $("#select_id ...
- find命令结合cp bash mv 命令使用的4种方式
工作经常需要用find结合其它命令一起使用,下面介绍4种结合方式. 例: 用find查找/data目录下,以.txt文件结尾的文件并复制到/tmp下 方法一 find与|xargs是黄金搭档,-t 参 ...
- 【PM日记】处理事务的逻辑
首先你得时刻搞清楚在你的当下什么类型事情是最重要的,是与人交流,是推进项目,还是需要更加埋头学习知识. 每天你得有个list,可以是上一日遗留下来的部分未完成项,可以是idea收集箱中拿到的新任务,总 ...
- eclipse 下安装 lombok.jar
lombok是一个java 开发插件,可以用来简化代码, 1. 下载lombok.jar https://projectlombok.org/download 2 将lombok.jar文件放在ecl ...