Ribosome profiling|N-terminomics|蛋白质基因组学
生物医学大数据-蛋白质基因组学:质谱注释
蛋白质组与其他组学的关系便是互为印证:蛋白质基因组学原本用于基因组注释,后面扩展到蛋白质与转录组或可变剪接之间关系,同时,蛋白质组依赖于基因组注释作为验证。许多研究未标明蛋白质基因组学,而是归属于对应的组学。
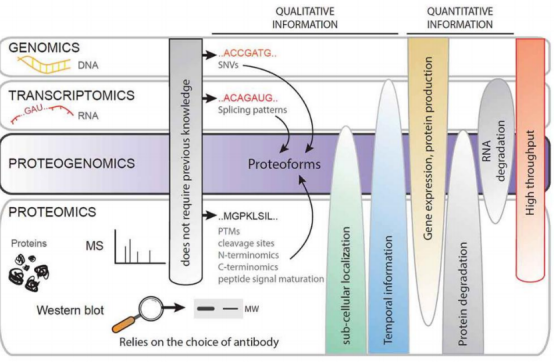
蛋白质基因组学现存问题:
基因组注释方法:1.Denovo。2.与转录组相应证。3.与基因组数据库同源比对。
基因组注释问题:
对于特定结构:
- 起始位点定位信号难以确定
- 确定promoter调控结合位点,错误率高
- 终止位点不被注释
- 可变剪接预测和确认上需要更多基因组的训练数据,使用denovo可以全部预测出来,但是用内含子和外显子仅能预测60%。
对于基因组的常规部分:
- 因为二代reads短所以难拼接
- 以前是鉴定已经存在的基因组,现在是未知物种的genome
- 同类数据不同处理有不同结果
- 人员水平不齐
对于蛋白质组大量质谱数据没被充分解析,可能的原因是
- 蛋白质多种修饰
- 搜索引擎差异
- 其它电荷没鉴定
- 数据库数据不足
没有充分解析造成许多没鉴定出来的数据,这些数据有可能是
- 噪音
- 实验污染,通过加入污染蛋白证实这部分是实验污染,当然鉴定出来之后也会舍弃这部分,对于该部分问题需要改进质控方法。
- 产生混合图谱(即多肽段在同一个图谱)或者融合肽段
- 在已知信息库中未收录的蛋白,即新蛋白
蛋白质基因组学的作用:
1.修正基因模型,即增加新注释,增加新肽段2.反过来由新肽段增加新基因
蛋白质基因组学需要提高的方法
蛋白质实验方面:
- 提高分离技术使蛋白质分的开
- 提高富集技术使得蛋白质量变大
- 高精度仪器
- 提高样本多样性,收集来源于不同时间空间的样本
数据库及数据处理方面:
常规部分存在的问题是大数据库存有大量噪音和相似,数据六位翻译结果搜索空间和denovo差不多。可以通过限制外显子大小,保留高分和正负库方法改进。质量评估时采用多段鉴定。假阳性高,动态范围宽,难鉴定可变剪接(可变剪接来源于denovo大数据库),重拼接假阳性高。
搜索速度方面:
质量过滤低质量、图谱聚类、去重或计算机并行的方法改良检索速度。
新方法:
加入RNAseq比对多重验证。
分成亚细胞组分多种方法鉴定。
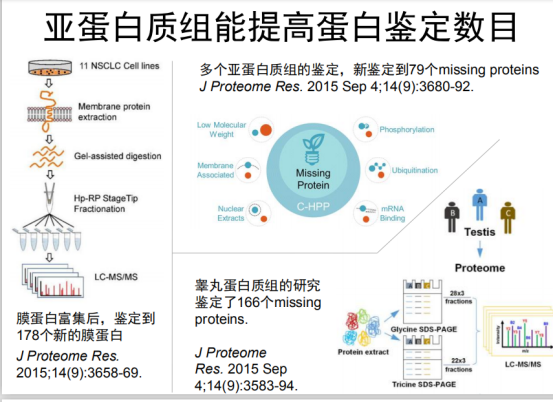
体外转录翻译来验证

n-末端组学:使用对角线色谱查肽段末端

DNA和RNA数据辅助蛋白质鉴定

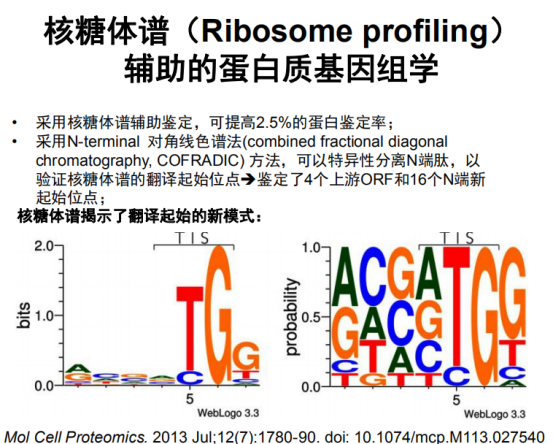
核糖体谱
Ribosome profiling|N-terminomics|蛋白质基因组学的更多相关文章
- 【蛋白质基因组】Proteogenomics方法介绍及分析思路
概念 利用蛋白质组学数据,结合基因组数据(DNA).转录组数据(RNA)来研究基因组注释问题,被称为蛋白质基因组学."蛋白质基因组学"一词由Jaffe 等于2004 年首次提出,作 ...
- lncRNA研究
------------------------------- Long noncoding RNAs are rarely translated in two human cell lines. ( ...
- dbSNP|n SwissVar|n CanProVar|CHPP|mutation assessor|
癌症蛋白质基因组学主要研究driver性质的突变,该突变有可能是转化为癌基因的突变.抑癌基因突变.药物位点突变和蛋白突变,可以使用mutation assessor 预测突变 突变导致疾病,修饰仅影响 ...
- ESTScan|EORF|Augustus|nr|PSM|
生物信息学方法的目的有二:1.常规找鉴定已知蛋白2.鉴定新蛋白 控制数据库大小可以通过增多酶切使得大数据库灵敏性增高数据量变小: 分步搜索是对于经典方法使用后找不到的新蛋白进行补充挖掘,预测蛋白与高可 ...
- Mol Cell Proteomics. | Integration and analysis of CPTAC proteomics data in the context of cancer genomics in the cBioPortal (解读人:徐洪凯)
文献名:Integration and analysis of CPTAC proteomics data in the context of cancer genomics in the cBioP ...
- 【Proteogenomis】工具评述
2015年的综述文章,marker一下,供以后比较试用.蛋白质基因组学研究中的质谱仪与生物信息学方法. 针对蛋白质基因组学的分析研究,总结了质谱仪和蛋白质基因组学流程分析软件的选择,重点评述了常用的生 ...
- DIP|PCN|CoevDB|PID|Y2H|RosettaDock Serve|元基因组学|微生物多样性
生命组学: 比较真核生物有关呼吸链的gene是比较核外编码基因,因为与呼吸有关的功能在线粒体上,线粒体位于核外.想要查看两种基因是否具有相互作用,可以对不同物种的编码ATP6 和ATP8的直系同源基因 ...
- Alterations of brain quantitative proteomics profiling revealed the molecular mechanisms of diosgenin against cerebral ischemia reperfusion effects(大脑的定量蛋白质组学揭示了薯蓣皂苷元对脑缺血再灌注效应的分子机制)
文献名:Alterations of brain quantitative proteomics profiling revealed the molecular mechanisms of dios ...
- Deep Protein Methylation Profiling by Combined Chemical and Immunoaffinity Approaches Reveals Novel PRMT1 Targets (结合层析法和免疫沉淀法的蛋白甲基化的深度检测技术发现了PRMT1新的靶标蛋白)
题目:Deep Protein Methylation Profiling by Combined Chemical and Immunoaffinity Approaches Reveals Nov ...
随机推荐
- 吴裕雄--天生自然 PHP开发学习:For 循环
<?php for ($i=1; $i<=5; $i++) { echo "The number is " . $i . "<br>"; ...
- github新建一个单页
比如可以在github上打开的网页是这种网址形式的:https://01xunsicheng.github.io/yumeihua/ 1.登录后首页找到 New repository 2.新建一个文件 ...
- Docker部署zookeeper集群和kafka集群,实现互联
本文介绍在单机上通过docker部署zookeeper集群和kafka集群的可操作方案. 0.准备工作 创建zk目录,在该目录下创建生成zookeeper集群和kafka集群的yml文件,以及用于在该 ...
- inotifywait命令详解及安装
https://www.cnblogs.com/pyrene/p/6414724.html 安装 https://www.cnblogs.com/martinzhang/p/4126907.h ...
- 1. react 基础 简介 及 环境搭建
一.简介 由 Facebook 推出 2013 年 开源 的 函数式编程的 使用人数最多的 前端框架 拥有健全的文档与完善的社区 ( 官网 ) react 16 称为 React Fiber ( 底层 ...
- 细说opcache
; opcache的开关,关闭时代码不再优化. opcache.enable=1 ; Determines if Zend OPCache is enabled for the CLI version ...
- 《Docekr入门学习篇》——Docker网络及数据卷
Docker网络设置 默认情况下docker会创建一个桥接网卡[docker 0],docker有两种映射方式,一种是随机映射,一种是指定映射. 提示:生产场景一般不使用随机映射,但是随机映射的好处是 ...
- 吴裕雄--天生自然ShellX学习笔记:Shell 基本运算符
Shell 和其他编程语言一样,支持多种运算符,包括: 算数运算符 关系运算符 布尔运算符 字符串运算符 文件测试运算符 原生bash不支持简单的数学运算,但是可以通过其他命令来实现,例如 awk 和 ...
- 三、VIP课程:并发编程专题->01-并发编程之Executor线程池详解
01-并发编程之Executor线程池详解 线程:什么是线程&多线程 线程:线程是进程的一个实体,是 CPU 调度和分派的基本单位,它是比进程更小的能独立运行的基本单位.线程自己基本上不拥有系 ...
- Linux-进程关系
(1).无关系 (2).父子进程关系 (3).进程组(group):由若干个进程构成一个进程组 (4).会话(session):由若干个进程组构成一个会话