Analyzing Microarray Data with R
1) 熟悉CEL file
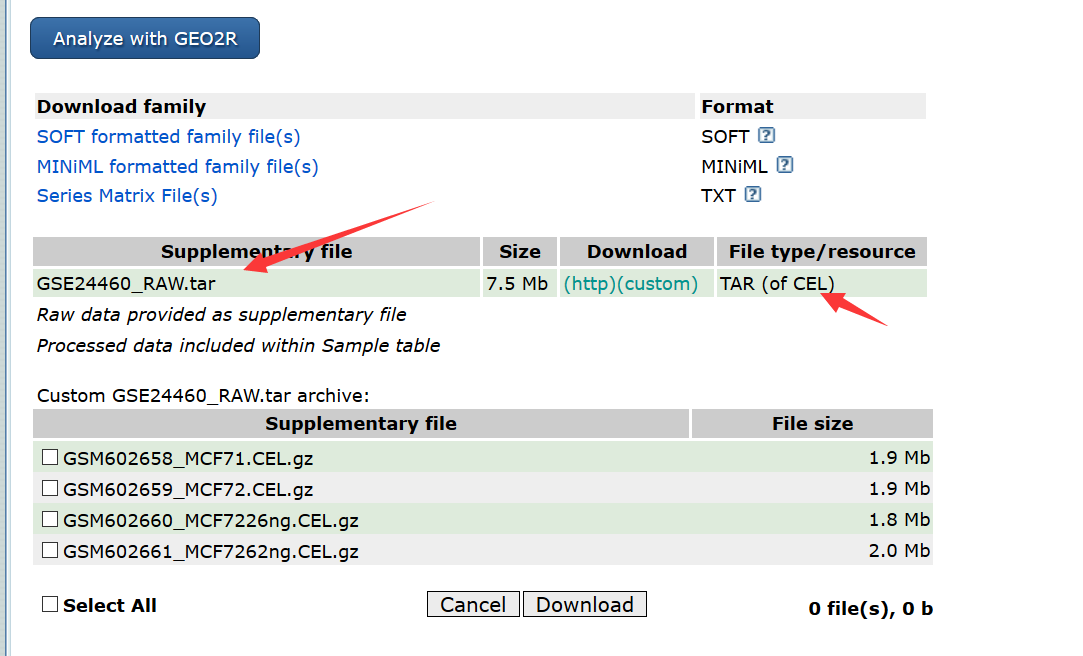
从 NCBI GEO (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE24460)下载GSE24460. 将得到一个 GSE24460_RAW.tar 文件,解压。产生CEL文件,包含各种信息。
if("affy" %in% rownames(installed.packages()) == FALSE) {source("http://bioconductor.org/biocLite.R");biocLite("affy")}
suppressMessages(library(affy))
ls('package:affy')
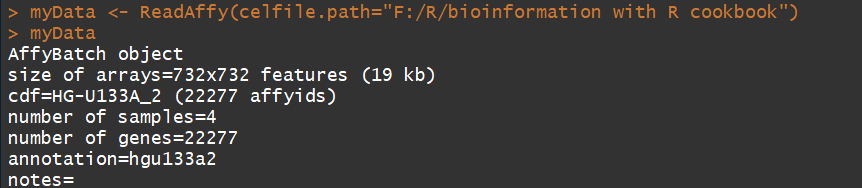
myData <- ReadAffy(celfile.path="F:/R/bioinformation with R cookbook") #ReadAffy()返回的是一个AffyBatch object(对象)
#myData1 <- ReadAffy(filenames = "F:/R/bioinformation with R cookbook/GSM602658_MCF71.CEL") #读取单个文件
--------如果不是从CEL文件读取,而是有多个独立的临床、实验、表达矩阵等文件,则需根据这些文件构建新的ExpressionSet对象,如下例子:---------------
每一部分组合成 ExpressionSet 对象,都扮演各自的角色。 exprs object 是表达量, phenotypic data 是样本临床信息 ( sex, age, treatment ...), annotated package 提供基本数据操作工具 。
##############################构建ExpressionSet对象(包含临床、实验、表达矩阵等多种信息)###############################
######利用自带数据集演示#######
suppressMessages(library(Biobase))
DIR <- system.file("extdata", package="Biobase")
exprsLoc <- file.path(DIR, "exprsData.txt")
pDataLoc <- file.path(DIR, "pData.txt") exprs <- as.matrix(read.csv(exprsLoc, header = TRUE, sep = "\t", row.names = 1, as.is = TRUE))#读取表达矩阵
class(exprs)
dim(exprs)
pData <- read.table(pDataLoc, row.names = 1, header = TRUE, sep = "\t") #读取临床信息
pData <- new("AnnotatedDataFrame", data = pData) #构建pData对象
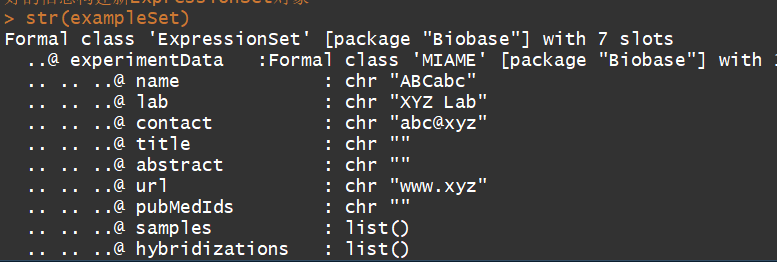
exData <- new("MIAME", name="ABCabc", lab="XYZ Lab", contact="abc@xyz", title="", abstract="", url="www.xyz") #编译实验信息,这个不是必须的
exampleSet <- new("ExpressionSet", exprs = exprs, phenoData = pData, experimentData = exData, annotation = "hgu133a2")#利用上边编译好的信息构建ExpressionSet对象
str(exampleSet)
validObject(exampleSet) #检验构建的ExpressionSet对象的有效性
2)Handling the AffyBatch object(了解affBatch对象结构)
myData
str(myData)
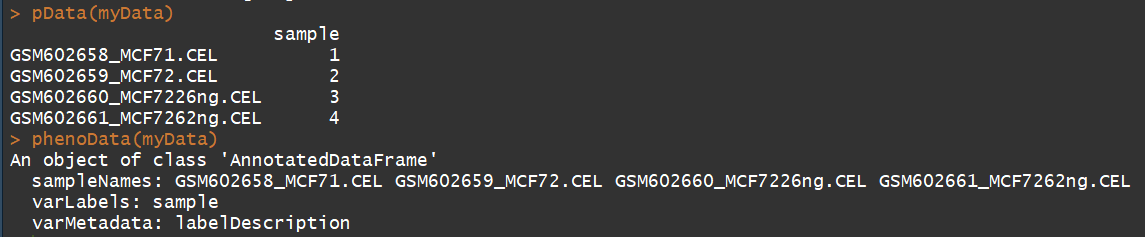
pData(myData) #临床信息
phenoData(myData)
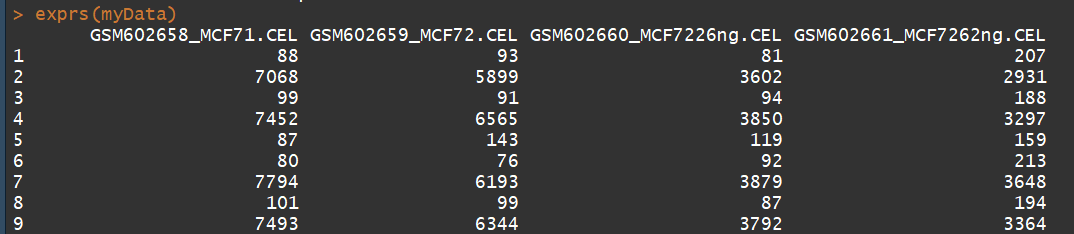
exprs(myData) #获取表达矩阵
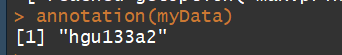
annotation(myData)# 获取注释信息
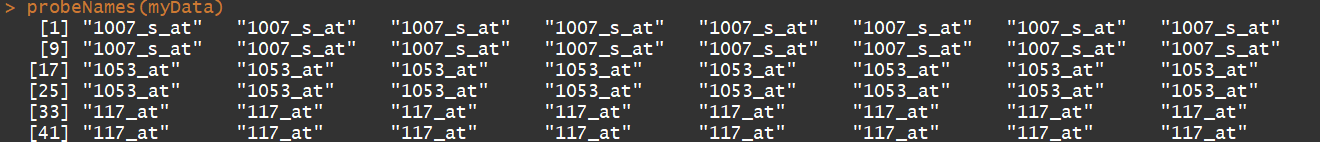
probeNames(myData) #获取探针名称
sampleNames(myData) #获取样本名称

3)Checking the quality of data(质控)
与质量相关的问题可能源于:
1、杂交,因为芯片上的荧光不均匀会导致不同的强度分布,非特异性结合或其他生物/技术原因可能在数据中产生背景噪声。
2、不合适的实验设计可能会影响整个数据集。 使用此类数据将导致数据分析期间的错误或不确定的推断。
因此,必须在开始数据分析之前确保数据质量。 这是通过寻找边远数组,数组内的分布,批处理效果等来实现。 有各种分析和诊断图可用于计算这些度量,以解释分析中的阵列数据的质量。
if("arrayQualityMetrics" %in% rownames(installed.packages()) == FALSE) {source("http://bioconductor.org/biocLite.R");biocLite("arrayQualityMetrics")}
suppressMessages(library(arrayQualityMetrics))
if("arrayQualityMetrics" %in% rownames(installed.packages()) == FALSE) {source("http://bioconductor.org/biocLite.R");biocLite("arrayQualityMetrics")}
suppressMessages(library(arrayQualityMetrics))
arrayQualityMetrics(myData, outdir="microarray") #质控
browseURL(file.path("microarray", "index.html"))
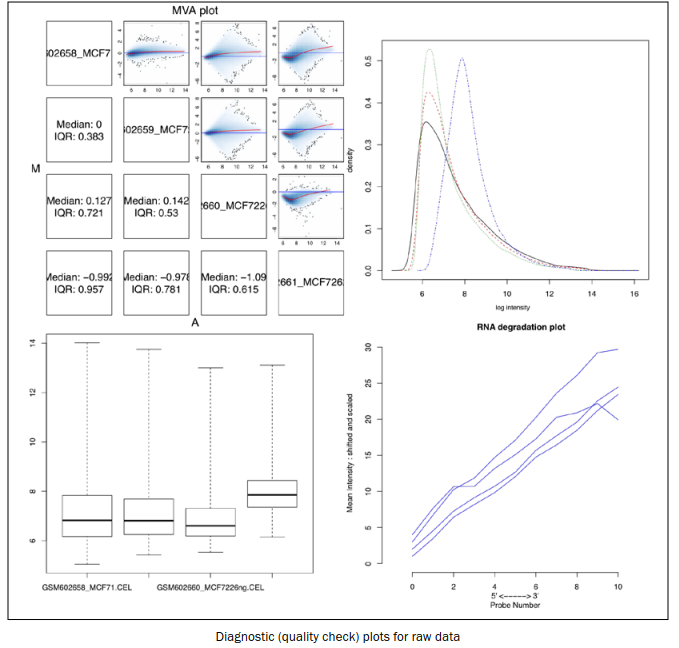
MAplot(myData, pairs=TRUE, plot.method="smoothScatter") #MAplot图
plotDensity.AffyBatch(myData) #密度图
boxplot(myData) #箱型图
rnaDeg <- AffyRNAdeg(myData) #查看RNA降解
plotAffyRNAdeg(rnaDeg)
summaryAffyRNAdeg(rnaDeg) #获取RNA降解情况
可以生成网页版报告:

4)Generating artificial expression data(仿真数据)
install.packages("madsim")
library(madsim)
fparams <- data.frame(m1 = 7, m2 = 7, shape2 = 4, lb = 4, ub =
14, pde = 0.02, sym = 0.5)#define your first set of parameters for the simulation process
dparams <- data.frame(lambda1 = 0.13, lambda2 = 2, muminde = 1,
sdde = 0.5) # Define the second set of parameters that consists of the statistical parameters
sdn <- 0.4
rseed <- 50
n <- 35000 #define the number of genes you require in the expression data
myData <- madsim(mdata=NULL, n=35000, ratio=0, fparams, dparams, sdn, rseed)#generate the synthetic data
str(myData)
library(limma)
plotMA(myData[[1]], 1) # visualize the data, create an MA plot for any sample, say, #sample 1,
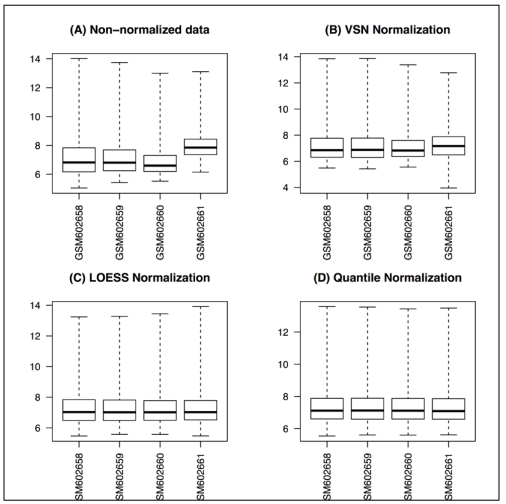
5)Data normalization
标准化用来降低技术影响,产生可比较数据。因为有多种方法可以标准化数据,这里讲vsn, loess, quantile三种方法。
library(vsn)
ls('package:vsn')
myData <- ReadAffy(celfile.path="F:/R/bioinformation with R cookbook") #读取所有CEL文件,返回的是AffyBatch object
#myData.VSN <- normalize.AffyBatch.vsn(myData) #vsn包提示找不到该函数
myData.loess <- normalize.AffyBatch.loess(myData)
boxplot(myData.loess)
myData.quantile <- normalize.AffyBatch.quantiles(myData)
boxplot(myData.quantile)
效果:
6)Overcoming batch effects in expression data
批次效应:是由于对样本不同批次的操作,属于实验样本间的非生物误差。产生原因包括sample preparation or hybridization protocol等,可以在一定程度间消除,但不可避免。因此需要对数据进行一定的预处理。
source("http://bioconductor.org/biocLite.R")
biocLite("bladderbatch")
library(sva) # contains batch removing utilities
library(bladderbatch) # The data to be used
data(bladderdata)
pheno <- pData(bladderEset) #提取表达矩阵 and pheno data
edata <- exprs(bladderEset) #提取样本临床信息
pheno
myData <- bladderEset[,sampleNames(bladderEset)[1:8]] #提取同样本不同批次子集
arrayQualityMetrics(myData, outdir="qc_be") #质控
mod1 <- model.matrix(~as.factor(cancer), data=pData(myData))[,c(1,3)] #model矩阵
batch <- pData(myData)$batch #样本批次信息
edata <- exprs(myData) #表达矩阵
combat_edata <- ComBat(dat=edata, batch=batch, mod=mod1, par.prior=TRUE)#经验贝叶斯方法去merge批次效应
myData2 <- myData
exprs(myData2) <- combat_edata
arrayQualityMetrics(myData2, outdir="qc_nbe")
批次效应如下左图,同一批次的样本进行聚类,移除批次效应之后,见右图
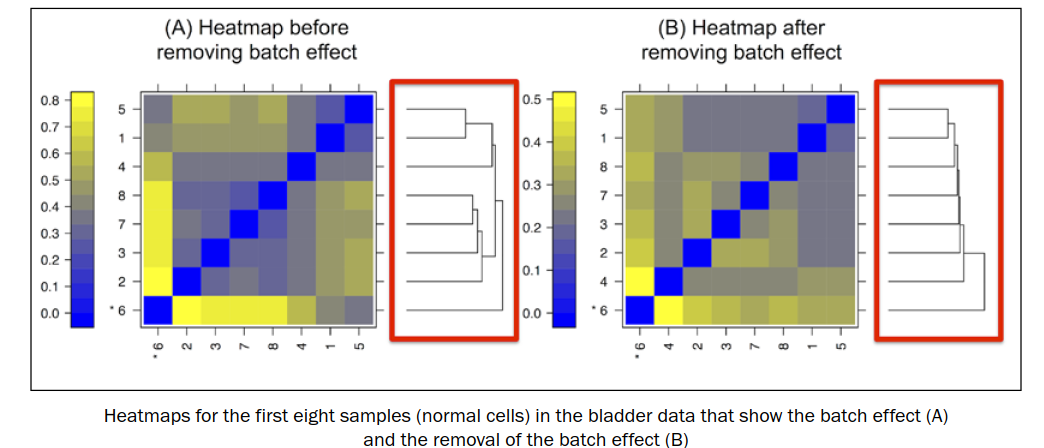
7)样本间关系(An exploratory analysis of data with PCA)
myData <- ReadAffy(celfile.path="F:/R/bioinformation with R cookbook") #读取所有CEL文件,返回的是AffyBatch object
myData.pca <- exprs(myData)
myPca <- prcomp(myData.pca, scale=TRUE) #prcomp函数计算主成分
summary(myPca)
colors <- c("green","cyan","violet","magenta") #给样本设计颜色
pairs(myPca$x, col=colors)
8)差异表达基因(Finding the differentially expressed genes)
library(affy) # Package for affy data handling
library(antiProfilesData) # Package containing input data
library(affyPLM) # Normalization package for eSet
library(limma) # limma analysis package
data(apColonData)
myData <- apColonData[, sampleNames(apColonData)[1:16]]
myData_quantile <- normalize.ExpressionSet.quantiles(myData)
design <- model.matrix(~0 + pData(myData)$Status)
fit <- lmFit(myData_quantile,design)
fit
fitE <- eBayes(fit)
tested <- topTable(fitE, adjust="fdr", sort.by="B", number=Inf)
DE <- x[tested$adj.P.Val<0.01,]
dim[DE]
DE <- x[tested$adj.P.Val< 0.01 & abs(x$logFC) >2,]
9)多组比较,最主要的是设计分组矩阵
library(leukemiasEset)
data(leukemiasEset)
pheno <- pData(leukemiasEset)
myData <- leukemiasEset[, sampleNames(leukemiasEset)[c(1:3, 13:15, 25:27, 49:51)]]
design <- model.matrix(~0 + factor(pData(myData)$LeukemiaType)) #分组矩阵
colnames(design) <- unique(as.character(pData(myData)$LeukemiaType))
design
fit <- lmFit(myData, design)
contrast.matrix <- makeContrasts(NoL- ALL, NoL- AML, NoL- CLL,
levels = design)
fit2 <- contrasts.fit(fit, contrast.matrix)
fit2 <- eBayes(fit2)
tested2 <- topTable(fit2,adjust="fdr",sort.by="B",number=Inf,
coef=1)
DE2 <- tested2[tested2$adj.P.Val < 0.01,]
dim(DE2)
10)Handling time series data
biocLite("Mfuzz")
library(Mfuzz)
biocLite("affyPLM")
library(affyPLM)
data(yeast)
plotDensity(yeast)
boxplot(yeast)
yeast_norm <- normalize.ExpressionSet.quantile(yeast)
pData(yeast_norm)
times <- pData(yeast_norm)$time
times <- as.factor(times)
design <- model.matrix(~0 +factor(pData(yeast_norm)$time))
colnames(design)[1:17] <- c("C", paste("T", 0:16, sep=""))
cont <- makeContrasts(C-T1, C-T2, C-T3, C-T4, C-T5, C-T6, C-T7,
C-T8, C-T9, C-T10, C-T11, C-T12, C-T13, C-T14, C-T15, C-T16,
levels=design)
fit <- lmFit(yeast_norm, cont)
fitE <- eBayes(fit)
x <- topTable(fitE, adjust="fdr", sort.by="F", number=100)
x[x$adj.P.Val< 0.05,]
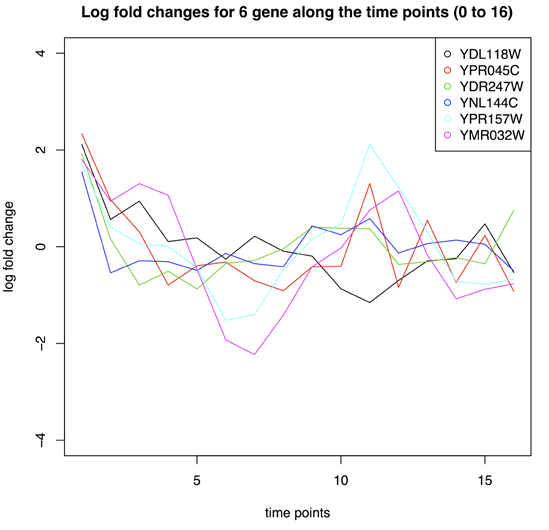
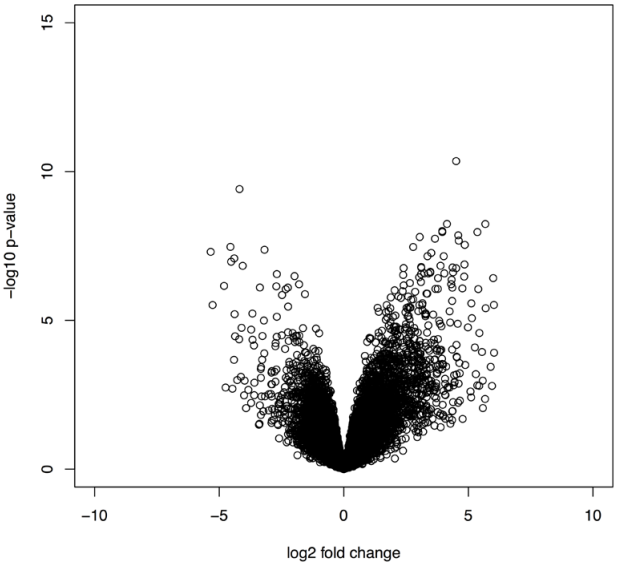
11)Fold changes in microarray data
head(DE2)
myTable <- topTable(fit, number=10000)
logratio <- tested2$logFC
library(gtools)
LR <- foldchange2logratio(foldchange, base=2)
FC <- logratio2foldchange(logratio, base=2)
plot(tested2$logFC, -log10(tested2$P.Value),xlim=c(-10, 10),
ylim=c(0, 15), xlab="log2 fold change", ylab="-log10 p-value")
myTable[tested2$P.Val< 0.05&logFC>1.5,]
12)The functional enrichment of data
Analyzing Microarray Data with R的更多相关文章
- (转) 6 ways of mean-centering data in R
6 ways of mean-centering data in R 怎么scale我们的数据? 还是要看我们自己数据的特征. 如何找到我们数据的中心? Cluster analysis with K ...
- Importing data in R 1
目录 Importing data in R 学习笔记1 flat files:CSV txt文件 packages:readr read_csv() read_tsv read_delim() da ...
- R0—New packages for reading data into R — fast
小伙伴儿们有福啦,2015年4月10日,Hadley Wickham大牛(开发了著名的ggplots包和plyr包等)和RStudio小组又出新作啦,新作品readr包和readxl包分别用于R读取t ...
- Cleaning Data in R
目录 R 中清洗数据 常见三种查看数据的函数 Exploring raw data 使用dplyr包里面的glimpse函数查看数据结构 \(提取指定元素 ```{r} # Histogram of ...
- Tutorial: Analyzing sales data from Excel and an OData feed
With Power BI Desktop, you can connect to all sorts of different data sources, then combine and shap ...
- Visualization data using R and bioconductor.--NCBI
- An Introduction to Stock Market Data Analysis with R (Part 1)
Around September of 2016 I wrote two articles on using Python for accessing, visualizing, and evalua ...
- Factoextra R Package: Easy Multivariate Data Analyses and Elegant Visualization
factoextra is an R package making easy to extract and visualize the output of exploratory multivaria ...
- Managing Spark data handles in R
When working with big data with R (say, using Spark and sparklyr) we have found it very convenient t ...
随机推荐
- 将DataTable 覆盖到 SQL某表(包括表结构及所有数据)
调用代码: string tableName = "Sheet1"; openFileDlg.ShowDialog(); DataTable dt = GeneralFun.Fil ...
- mysql 按照 where in 排序
select * from user_extend where `unique` in('mark.liu@xxxx.com','jason.gan@xxxx.com','ssgao@xxxx.com ...
- 关于lidroid xUtils 开源项目
最近搜了一些框架供初学者学习,比较了一下XUtils是目前git上比较活跃 功能比较完善的一个框架,是基于afinal开发的,比afinal稳定性提高了不少,下面是介绍: xUtils简介 xUtil ...
- bzoj1196 公路修建问题
Description OI island是一个非常漂亮的岛屿,自开发以来,到这儿来旅游的人很多.然而,由于该岛屿刚刚开发不久,所以那里的交通情况还是很糟糕.所以,OIER Association组织 ...
- Maven报错找不到jre
常规配置maven环境变量,报错: The JAVA_HOME environment variable is not defined correctly. This environment vari ...
- java如何写自己的native方法实现调用本地的c++库?
等待编辑 1:首先可以找一本jni java native interface相关的书籍来看.
- Spark Standalone模式HA环境搭建
Spark Standalone模式常见的HA部署方式有两种:基于文件系统的HA和基于ZK的HA 本篇只介绍基于ZK的HA环境搭建: $SPARK_HOME/conf/spark-env.sh 添加S ...
- 基于Linux的Samba开源共享解决方案测试(六)
在极限读场景下,对于客户端的网络监控如图: 在极限写场景下,对于NAS1网关的网络监控如图: 在极限写场景下,对于NAS2网关的网络监控如图: 在极限写场景下,对于客户端的网络监控如图: 在极限混合读 ...
- lambda,sorted(),filter(),map(),递归,二分法
1. lambda 匿名函数 语法: lambda 参数:返回值 不能完成复杂的操作例 # li=['21','asdd','weqeqw','wqf']# # it=iter(li)# # prin ...
- Dep数据发布,推送
package com.cfets.ts.u.shchgateway.util; import com.cfets.cwap.s.stp.MessageUnit; import com.cfets.t ...