Mol Cell Proteomics. | Prediction of LC-MS/MS properties of peptides from sequence by deep learning (通过深度学习技术根据肽段序列预测其LC-MS/MS谱特征) (解读人:梅占龙)
通过深度学习技术根据肽段序列预测其LC-MS/MS谱特征
解读人:梅占龙 质谱平台
文献名:Prediction of LC-MS/MS properties of peptides from sequence by deep learning
期刊名:Molecular & Cellular Proteomics
发表时间:2019年9月
IF:4.828
作者:
Shenheng Guan 1,2,* , Michael F. Moran 2,3 , and Bin Ma 1
单位:
1加拿大滑铁卢大学滑铁卢大学戴维·R·切里顿计算机科学学院,加拿大N2L 3G1
2儿童医院细胞生物学和SPARC生物中心, 安大略省多伦多市湾街686号, 加拿大,M5G 0A4
3多伦多大学分子遗传学系,安大略省多伦多市Bay Bay 686号,M5G 0A4,加拿大
一、 概述:
本文开发了根据肽段序列预测来三个关键LC-MS/MS特性的深度学习模型。 LC-MS/MS的特性指的是保留时间(iRT),MS1电荷分布以及HCD谱图的子离子强度分布。利用核心深度监督学习体系结构,双向长期短期记忆(LSTM)递归神经网络来构建这三个预测模型。本文提出并展示了两种个性化方案以对修饰进行分析。使用2 X 106实验谱图对HCD碎裂谱图预测模型进行了训练。与文献中可用的最新模型相比,iRT预测模型和HCD谱图预测模型的准确性更高。 MS1电荷分布预测模型预测性能更出色。预测模型可用于在数据依赖采集(DDA)和数据非依赖采集(DIA)实验中增强肽段的鉴定和定量准确性,以及协助MRM和PRM实验设计。
二、 研究背景:
从肽段序列直接预测LC-MS/MS特性的能力将为下一代蛋白质组学研究提供助力。当前,肽段的主要行为或性质是通过实验获得的,成本高,准确性也有较大差异。这项工作旨在使用经过实验数据训练得到的深度学习模型来解决这些问题,以预测有用的LC-MS/MS特性,例如保留时间(iRT),MS1电荷状态分布以及MS/MS子离子强度。
从本世纪初开始,基于神经网络的深度学习模型对图像和自然语言处理产生了重大影响。对于肽段属性预测,循环神经网络模型(例如长期短期记忆(LSTM)模型)深度学习都是不错的选择,因为它们能够处理长度可变的输入数据,例如肽段序列。深度监督学习模型擅长处理分类和回归任务。我们的iRT和谱图预测模型是经典的回归模型,但电荷分布预测模型也是一种分类方法,其输出为电荷分布概率。这里展示的三个预测模型代表LC-MS/MS中的三个特征:用于表征LC行为的iRT,用于MS1的电荷分布以及MS/MS级别的HCD子离子强度。它们的输出维数也不同:iRT是标量,电荷状态分布是矢量,HCD子离子强度是二维矩阵。核心学习模块采用的是双向LSTM层,这保障了深度学习模型在各种问题上的适用性。肽段特性预测模型的性能主要取决于大量和高质量的训练数据。我们的HCD子离子强度预测模型的训练集就是从发表的研究中收集的大量高质量谱图。
三、 实验方法:
模型构建:所有模型都由一个掩膜层(masking layer)和两个双向的长期短期记忆(LSTM)层组成。为了得到无偏的模型,在训练集的建立中,应用了有放回和无放回取样。三个模型在输出层上有所不同:对于iRT模型,输出为标量,使用了两个具有双曲正切函数的致密层,最终输出的维度为1。对于电荷分布模型,存在两个密集层,其输出为长度为5的一维数组(对应于电荷状态1至5)。对于谱图预测模型,主要使用时间分布密集层。所有使用Keras框架实现的LSTM模型都在带有11GB显存的GeForce RTX 2080 Ti显卡上进行训练。 HCD子离子预测模型的一个子集(1074898谱图)的最长训练时间为7.2小时。
四、研究成果:
1. 建立了三个深度学习模型,用以预测肽段的三个关键LC-MS/MS属性:保留时间(iRT),MS1电荷分布和HCD子离子强度。 这三个特性在三个不同的实验水平上表征了肽段的行为:色谱,MS1以及MS/MS水平。
2. 如图1,2所示,iRT模型的预测效果较为准确。其中有一些异常点可能是错误的实验鉴定结果引起的。与DeepRT模型的预测相比,该模型iRT预测的准确度高约28%。 iRT预测的95%置信区间几乎比DeepRT的小两倍。
3. 对于电荷分布预测,预测得到的电荷分布与实验得到的结果,其皮尔逊相关系数(PCC)中位数为0.997(图3)。并且该模型还可以进行常见的肽段修饰的设置(图4)。
4. 如图5所示,在训练集和测试集中,预测的子离子强度分布与实验得到的子离子强度分布的皮尔逊相关系数(PCC)分布非常相似,表明该模型并没有过拟合(若训练集的结果远好于测试集则可能过拟合了)。训练和测试集的PCC中位数分别为0.955和0.953。 这个测试数据集的皮尔逊系数中值PPC略好于pDeep得到的结果。
6. 提供了用于LC-MS/MS属性预测的Web服务。 可以从Zenodo(附件S6部分)下载经过训练的模型,训练和测试数据以及相关的python代码。
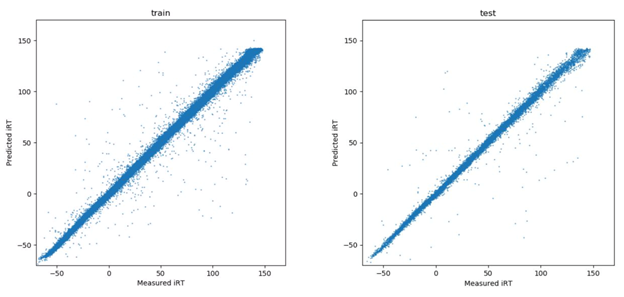
图1.测得的iRT与预测的iRT的散点图。左:训练数据集。右:测试数据集。
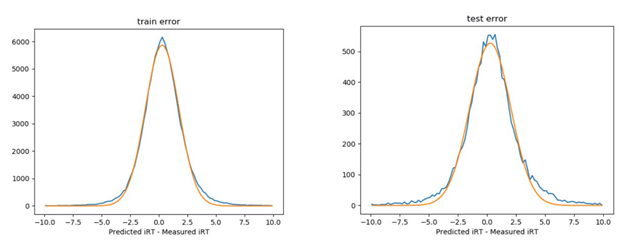
图2. iRT预测模型的误差分布。蓝色曲线是误差分布,橙色曲线是相应的高斯拟合。左:训练错误。右:测试错误。
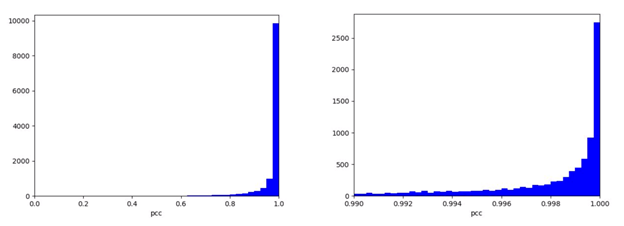
图3.电荷分布预测与测试数据集实验测量的结果的皮尔逊相关系数分布。用126,876个实验得到的电荷分布中的90%来训练MS1电荷分布预测模型。其中10%被用作测试数据集以评估该图所示的模型。左:全范围。右:[0.99,1]的放大范围。

图4.肽VLPGMHHPIQMKPADSEK的电荷分布预测(右)及其Q酰胺化形式(左)。上面的柱子(蓝色)是测量数据,下面的柱子(绿色)是预测值。
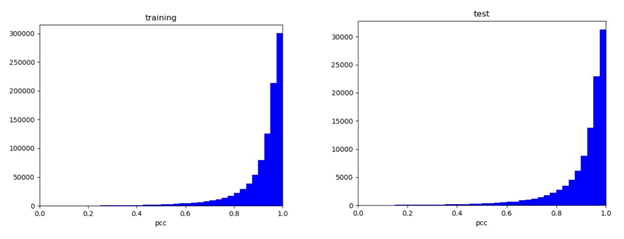
图5.实验和预测的HCD子离子强度之间的皮尔逊相关系数分布。左:训练数据集的预测结果;右:测试数据集预测结果。
阅读人:梅占龙
原文链接:https://www.mcponline.org/content/early/2019/06/27/mcp.TIR119.001412
DOI:https://doi.org/10.1074/mcp.TIR119.001412
Mol Cell Proteomics. | Prediction of LC-MS/MS properties of peptides from sequence by deep learning (通过深度学习技术根据肽段序列预测其LC-MS/MS谱特征) (解读人:梅占龙)的更多相关文章
- Mol Cell Proteomics. | Elevated Hexokinase II Expression Confers Acquired Resistance to 4-Hydroxytamoxifen in Breast Cancer Cells(升高的己糖激酶II表达使得乳腺癌细胞获得对他莫昔芬的抗性)(解读人:黄旭蕾)
文献名:Elevated Hexokinase II Expression Confers Acquired Resistance to 4-Hydroxytamoxifen in Breast Ca ...
- Mol Cell Proteomics. | 用于鉴定新型融合转录本及其在癌细胞中的潜在翻译产物的多功能蛋白质组基因组学工具FusionPro
期刊:Molecular & Cellular Proteomics 发表时间:June 17, 2019 DOI:10.1074/mcp.RA119.001456 分享人:任哲 内容与观点: ...
- Mol Cell Proteomics. |胡丹丹| 雷公藤红素通过SIRT1-FXR 信号通路保护胆汁淤积性肝损伤
期刊:Mol Cell Proteomics 题目:Celastrol protects from cholestatic liver injury though modulation of SIRT ...
- Mol Cell Proteomics. | 粪便微生物蛋白质的组成与饮食诱导肥胖倾向的关联研究
题目:Associations of the Fecal Microbial Proteome Composition and Proneness to Diet-induced Obesity 期刊 ...
- Mol Cell Proteomics. | MARMoSET – Extracting Publication-ready Mass Spectrometry Metadata from RAW Files
本文是马克思普朗克心肺研究所的三名研究者Marina Kiweler.Mario Looso和Johannes Graumann发表在8月刊的MCP的一篇文章. 由于Omics实验经常涉及数百个数据文 ...
- Mol. Cell. Proteomics | 糖蛋白基因组学:一种常见的基因多态性影响人血清胎球蛋白/α-2-HS-糖蛋白的糖基化形式
大家好,本次分享的是发表在Molecular & Cellular Proteomics上的一篇关于糖蛋白基因组学的文章,题目是Glycoproteogenomics: A Frequent ...
- Mol Cell Proteomics. | Proteomics Analysis of Extracellular Matrix Remodeling During Zebrafish Heart Regeneration (解读人:徐宁)
文献名:Proteomics Analysis of Extracellular Matrix Remodeling During Zebrafish Heart Regeneration(斑马鱼心脏 ...
- Mol Cell Proteomics. |王欣然| 基于微粒的蛋白聚合物捕获技术让能满足多种不同需求的蛋白质组学样品制备方法成为可能
大家好,本周分享的是发表在Molecular & Cellular Proteomics. 上的一篇关于蛋白质组学样本质谱分析前处理方法改进的文章,题目是Protein aggregation ...
- Mol Cell Proteomics. | A Targeted Mass Spectrometry Strategy for Developing Proteomic Biomarkers: A Case Study of Epithelial Ovarian Cancer(利用靶向质谱策略进行上皮性卵巢癌病例的蛋白质组生物标志物研究) (解读人:王聚)
文献名:利用靶向质谱策略进行上皮性卵巢癌病例的蛋白质组生物标志物研究 期刊名:Molecular & Cellular Proteomics 发表时间:(2019年7月) IF:5.41 单位 ...
随机推荐
- 使用face_recognition进行人脸特征检测
效果图调用face_recognition.face_landmarks()方法即可得到人脸特征点, 返回一个字典, 下图是返回的数据, 包括chin(下巴), left_eye(左眼)等.我画了两种 ...
- Python---13面向对象编程
一.类和实例 面向对象最重要的概念就是类(Class)和实例(Instance),必须牢记类是抽象的模板,比如Student类,而实例是根据类创建出来的一个个具体的“对象”,每个对象都拥有相同的方法, ...
- 剑指CopyOnWriteArrayList
上期回顾 之前的一篇 剑指ConcurrentHashMap[基于JDK1.8] 给大家详细分析了一波JUC的ConcurrentHashMap,它在线程安全的基础上提供了更好的写并发能力.那么既然有 ...
- 手术Robot能取代医生吗?
现在,机械自动化已经成为各领域为之神往的大趋势,从工业组装,到智能物流,再到餐饮.银行服务,以及娱乐等等,管理者无不处心积虑地降低成本.提高效率,其中,一个非常重要的手段就是利用机器取代人工.医院,作 ...
- loadrunner通过web的post请求方法测接口 2
模拟APP发送请求给Cloud, 一般都是用户登录后.cloud会返回登录成功的消息并且返回一个cookie给app, app下次要做一些例如设置名称之类的工作,在请求消息里面会携带返回的cookie ...
- 用Express 4和 MongoDB打造Todo List
本篇文章主要參考自DreamersLab - 用Express和MongoDB寫一個todo list.原文的教學內容是使用Express 3.x版,在這邊做簡單Express 4.12.1的todo ...
- angular知识点(2)
angular知识点(2) 1.为了代码规范,对于需要自动加载的依赖,需要在前面加上注释,注释为://@ngInject 或者是/*@ngInject*/ 2.ngSwitch的应用 在需要用到选择出 ...
- k8s集群搭建(三)
Dashboard安装 Kubernetes Dashboard是k8s提供基于Web的监控和操作界面,可以通过UI来显示集群的所有工作负载,除了查看资源,还是创建.编辑.更新.删除资源. 根据Kub ...
- AIDLservice
有三种情况:如果直接使用服务,则没有必要进行绑定,但是如果要使用服务里面的方法,则要进行绑定.具体的启动情况有下: 其中很重要一点:bindService和unbindService是成对出现的. 1 ...
- 前端JS题
题目如下: function Foo() { getName = function () { alert (1); }; return this; } Foo.getName = function ( ...