单细胞分析实录(8): 展示marker基因的4种图形(一)
今天的内容讲讲单细胞文章中经常出现的展示细胞marker的图:tsne/umap图、热图、堆叠小提琴图、气泡图,每个图我都会用两种方法绘制。
使用的数据来自文献:Single-cell transcriptomics reveals regulators underlying immune cell diversity and immune subtypes associated with prognosis in nasopharyngeal carcinoma. 去年7月发表在Cell Research上的关于鼻咽癌的文章,数据下载:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150430
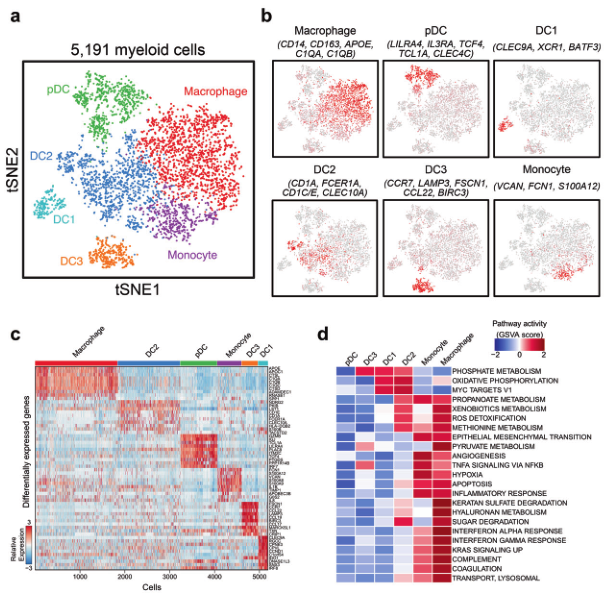
髓系细胞数量相对少一些,为了方便演示,选它作为例子。
library(Seurat)
library(tidyverse)
library(harmony)
Myeloid=read.table("Myeloid_mat.txt",header = T,row.names = 1,sep = "\t",stringsAsFactors = F)
Myeloid_anno=read.table("Myeloid_anno.txt",header = T,sep = "\t",stringsAsFactors = F)
导入数据的时候需要注意一个地方:从cell ranger得到的矩阵,每一列的列名会在CB后面加上"-1"这个字符串,在R里面导入数据时,会自动转化为".1",在做匹配的时候需要注意一下。我已经提前转换为"_1"
> summary(as.numeric(Myeloid["CD14",]))
Min. 1st Qu. Median Mean 3rd Qu. Max.
0.000 0.000 0.689 1.080 2.111 4.500
> summary(as.numeric(Myeloid["PTPRC",]))
Min. 1st Qu. Median Mean 3rd Qu. Max.
0.000 1.200 1.681 1.607 2.124 3.520
考虑到下载的表达矩阵,表达值都不是整数,且大于0,推测该矩阵已经经过了标准化,因此下面的流程会跳过这一步
0. Seurat流程
我们直接把注释结果赋值到mye.seu@meta.data矩阵中,后面省去聚类这一步
mye.seu=CreateSeuratObject(Myeloid)
mye.seu@meta.data$CB=rownames(mye.seu@meta.data)
mye.seu@meta.data=merge(mye.seu@meta.data,Myeloid_anno,by="CB")
rownames(mye.seu@meta.data)=mye.seu@meta.data$CB
#替代LogNormalize这一步
mye.seu[["RNA"]]@data=mye.seu[["RNA"]]@counts
mye.seu <- FindVariableFeatures(mye.seu, selection.method = "vst", nfeatures = 2000)
mye.seu <- ScaleData(mye.seu, features = rownames(mye.seu))
mye.seu <- RunPCA(mye.seu, npcs = 50, verbose = FALSE)
mye.seu=mye.seu %>% RunHarmony("sample", plot_convergence = TRUE)
mye.seu <- RunUMAP(mye.seu, reduction = "harmony", dims = 1:20)
mye.seu <- RunTSNE(mye.seu, reduction = "harmony", dims = 1:20)
#少了聚类
DimPlot(mye.seu, reduction = "tsne", group.by = "celltype", pt.size=1)+theme(
axis.line = element_blank(),
axis.ticks = element_blank(),axis.text = element_blank()
)
ggsave("tsne1.pdf",device = "pdf",width = 17,height = 14,units = "cm")
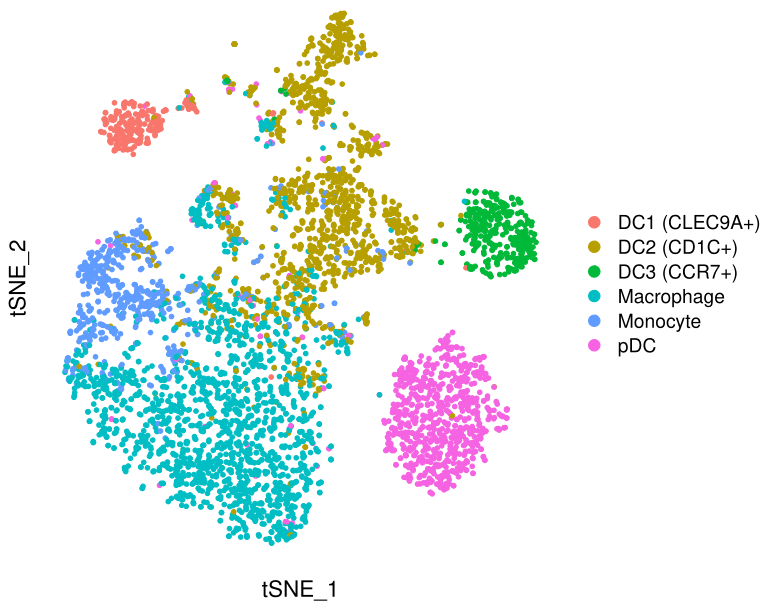
基本符合原图,三个亚群分得开,三个亚群分不开。
接下来,我用4种方式展示marker基因,这些基因可以在文献的补充材料里面找到。
1. tsne展示marker基因
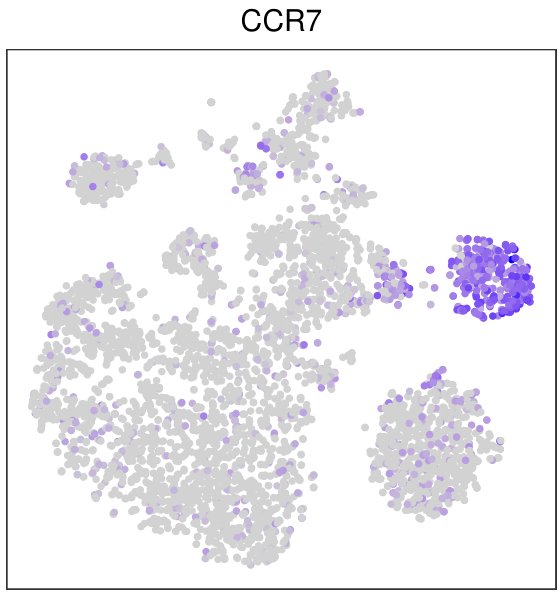
FeaturePlot(mye.seu,features = "CCR7",reduction = "tsne",pt.size = 1)+
scale_x_continuous("")+scale_y_continuous("")+
theme_bw()+ #改变ggplot2的主题
theme( #进一步修改主题
panel.grid.major = element_blank(),panel.grid.minor = element_blank(), #去掉背景线
axis.ticks = element_blank(),axis.text = element_blank(), #去掉坐标轴刻度和数字
legend.position = "none", #去掉图例
plot.title = element_text(hjust = 0.5,size=14) #改变标题位置和字体大小
)
ggsave("CCR7.pdf",device = "pdf",width = 10,height = 10.5,units = "cm")
另一种方法就是把tsne的坐标和基因的表达值提取出来,用ggplot2画,其实不是很必要,因为FeaturePlot也是基于ggplot2的,我还是演示一下
mat1=as.data.frame(mye.seu[["RNA"]]@data["CCR7",])
colnames(mat1)="exp"
mat2=Embeddings(mye.seu,"tsne")
mat3=merge(mat2,mat1,by="row.names")
#数据格式如下:
> head(mat3)
Row.names tSNE_1 tSNE_2 exp
1 N01_AAACGGGCATTTCAGG_1 5.098727 32.748145 0.000
2 N01_AAAGATGCAATGTAAG_1 -24.394040 26.176422 0.000
3 N01_AACTCAGGTAATAGCA_1 11.856730 8.086553 0.000
4 N01_AACTCAGGTCTTCGTC_1 10.421878 12.660407 0.000
5 N01_AACTTTCAGGCCATAG_1 33.555756 -10.437406 1.606
6 N01_AAGACCTTCGAATGGG_1 -23.976967 11.897753 0.738
mat3%>%ggplot(aes(tSNE_1,tSNE_2))+geom_point(aes(color=exp))+
scale_color_gradient(low = "grey",high = "purple")+theme_bw()
ggsave("CCR7.2.pdf",device = "pdf",width = 13.5,height = 12,units = "cm")

用ggplot2的好处就是图形修改很方便,毕竟ggplot2大家都很熟悉
2. 热图展示marker基因
画图前,需要给每个细胞一个身份,因为我们跳过了聚类这一步,此处需要手动赋值
Idents(mye.seu)="celltype"
library(xlsx)
markerdf1=read.xlsx("ref_marker.xlsx",sheetIndex = 1)
markerdf1$gene=as.character(markerdf1$gene)
# 这个表格整理自原文的附表,选了53个基因
#数据格式
# > head(markerdf1)
# gene celltype
# 1 S100B DC2(CD1C+)
# 2 HLA-DQB2 DC2(CD1C+)
# 3 FCER1A DC2(CD1C+)
# 4 CD1A DC2(CD1C+)
# 5 PKIB DC2(CD1C+)
# 6 NDRG2 DC2(CD1C+)
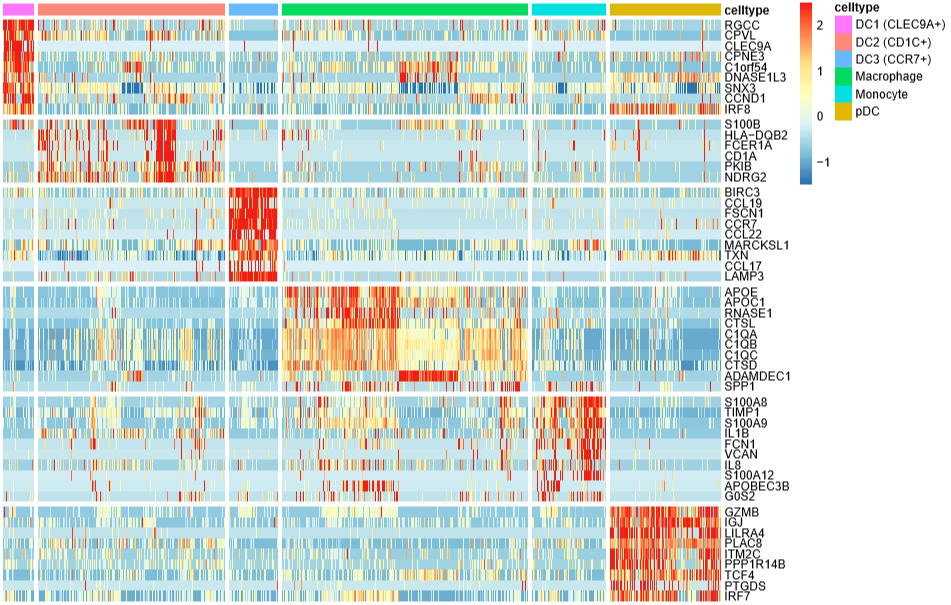
DoHeatmap(mye.seu,features = markerdf1$gene,label = F,slot = "scale.data")
ggsave("heatmap.pdf",device = "pdf",width = 23,height = 16,units = "cm")
label = F不在热图的上方标注细胞类型,
slot = "scale.data"使用scale之后的矩阵画图,默认就是这个

接下来用pheatmap画,在布局上可以自由发挥
library(pheatmap)
colanno=mye.seu@meta.data[,c("CB","celltype")]
colanno=colanno%>%arrange(celltype)
rownames(colanno)=colanno$CB
colanno$CB=NULL
colanno$celltype=factor(colanno$celltype,levels = unique(colanno$celltype))
先对细胞进行排序,按照celltype的顺序,然后对基因排序
rowanno=markerdf1
rowanno=rowanno%>%arrange(celltype)
提取scale矩阵的行列时,按照上面的顺序
mat4=mye.seu[["RNA"]]@scale.data[rowanno$gene,rownames(colanno)]
mat4[mat4>=2.5]=2.5
mat4[mat4 < (-1.5)]= -1.5 #小于负数时,加括号!
下面就是绘图代码了,我加了分界线,使其看上去更有区分度
pheatmap(mat4,cluster_rows = F,cluster_cols = F,
show_colnames = F,
annotation_col = colanno,
gaps_row=as.numeric(cumsum(table(rowanno$celltype))[-6]),
gaps_col=as.numeric(cumsum(table(colanno$celltype))[-6]),
filename="heatmap.2.pdf",width=11,height = 7
)
先写到这儿吧(原本以为能写完的),剩下的气泡图、堆叠小提琴图改天再补上。
因水平有限,有错误的地方,欢迎批评指正!
单细胞分析实录(8): 展示marker基因的4种图形(一)的更多相关文章
- 单细胞分析实录(9): 展示marker基因的4种图形(二)
在上一篇中,我已经讲解了展示marker基因的前两种图形,分别是tsne/umap图.热图,感兴趣的读者可以回顾一下.这一节我们继续学习堆叠小提琴图和气泡图. 3. 堆叠小提琴图展示marker基因 ...
- 【代码更新】单细胞分析实录(20): 将多个样本的CNV定位到染色体臂,并画热图
之前写过三篇和CNV相关的帖子,如果你做肿瘤单细胞转录组,大概率看过: 单细胞分析实录(11): inferCNV的基本用法 单细胞分析实录(12): 如何推断肿瘤细胞 单细胞分析实录(13): in ...
- 【代码更新】单细胞分析实录(21): 非负矩阵分解(NMF)的R代码实现,只需两步,啥图都有
1. 起因 之前的代码(单细胞分析实录(17): 非负矩阵分解(NMF)代码演示)没有涉及到python语法,只有4个python命令行,就跟Linux下面的ls grep一样的.然鹅,有几个小伙伴不 ...
- 单细胞分析实录(5): Seurat标准流程
前面我们已经学习了单细胞转录组分析的:使用Cell Ranger得到表达矩阵和doublet检测,今天我们开始Seurat标准流程的学习.这一部分的内容,网上有很多帖子,基本上都是把Seurat官网P ...
- 单细胞分析实录(4): doublet检测
最近Cell Systems杂志发表了一篇针对现有几种检测单细胞测序doublet的工具的评估文章,系统比较了常见的例如Scrublet.DoubletFinder等工具在检测准确性.计算效率等方面的 ...
- 单细胞分析实录(3): Cell Hashing数据拆分
在之前的文章里,我主要讲了如下两个内容:(1) 认识Cell Hashing:(2): 使用Cell Ranger得到表达矩阵.相信大家已经知道了cell hashing与普通10X转录组的差异,以及 ...
- 单细胞分析实录(18): 基于CellPhoneDB的细胞通讯分析及可视化 (上篇)
细胞通讯分析可以给我们一些细胞类群之间相互调控/交流的信息,这种细胞之间的调控主要是通过受配体结合,传递信号来实现的.不同的分化.疾病过程,可能存在特异的细胞通讯关系,因此阐明这些通讯关系至关重要. ...
- 单细胞分析实录(19): 基于CellPhoneDB的细胞通讯分析及可视化 (下篇)
在上一篇帖子中,我介绍了CellPhoneDB的原理.实际操作,以及一些值得注意的地方.这一篇继续细胞通讯分析的可视化. 公众号后台回复20210723获取本次演示的测试数据,以及主要的可视化代码. ...
- 单细胞分析实录(2): 使用Cell Ranger得到表达矩阵
Cell Ranger是一个"傻瓜"软件,你只需提供原始的fastq文件,它就会返回feature-barcode表达矩阵.为啥不说是gene-cell,举个例子,cell has ...
随机推荐
- deepFM(原理和pytorch理解)
参考(推荐):https://blog.csdn.net/w55100/article/details/90295932 要点: 其中的计算优化值得注意 K代表隐向量维数 n可以代表离散值one-ho ...
- web前端页面常见优化方法
(1)减少http请求,尽量减少向服务器的请求数量 (2)避免重定向 (3)利用缓存:使用外联式引用CSS.JS,在实际应用中使用外部文件可以提高页面速度,因为JavaScript和CSS文件都能在浏 ...
- react路由初探(1)
import React from 'react'; import logo from './logo.svg'; import './App.css'; class About extends Re ...
- Panda Global获悉,美国承诺4年内明确区块链数字资产监管方式!
近日,美国商品期货交易委员会(CFTC)宣布,在4年内将会全面把加密货币监管列为优先事项.Panda Global从7月8日公布的新战略中获悉,此次CFTC公布了自己接下来的新框架,并且在框架中承诺: ...
- AcWing 400. 太鼓达人
大型补档计划 题目链接 神仙题.考虑转为图论模型. 若以 \(2 ^ k\) 个点,相互转化,很容易看出要求一个哈密尔顿环,显然对于 \(1000\) 规模的数据求不出来. 对于图论中环的算法,并且能 ...
- http请求user_agent字段解析
浏览器的常见User Agent 各字段的解释 浏览器的User Agent字段令人迷惑,例如:某一版本的Chrome访问网络时,User Agent字段如下: Mozilla/5.0 (Window ...
- 使用 open 函数 写的代码 用户名登录
先创建文件ha.log 内容: aaa$$123bbb$$456 def dl(user,pas): f = open('ha.log', 'r', encoding="utf-8" ...
- MVCAdmin项目知识点记录
1.在过滤器中,用ViewBag类似的东西,要((ViewResult)filterContext.Result).ViewBag. 2.Controller中自己定义的非Action方法中(包括构造 ...
- sqli-labs less-24(二次注入)
less-24 原理: 在网站处理用户提交的数据的时候,只是将某些敏感字符进行了转义.因而使得用户第一次提交的时候不会被当做代码执行.但是这些数据存入数据库的时候却没有转义,而网站程序默认数据库中的数 ...
- 新手入门 : Windows Phone 8.1 开发 视频学习地址
本视频资源来自Microsoft Virtual Academy http://www.microsoftvirtualacademy.com/ 下面为视频下载地址! 新手入门 : Windows P ...