单细胞分析实录(18): 基于CellPhoneDB的细胞通讯分析及可视化 (上篇)
细胞通讯分析可以给我们一些细胞类群之间相互调控/交流的信息,这种细胞之间的调控主要是通过受配体结合,传递信号来实现的。不同的分化、疾病过程,可能存在特异的细胞通讯关系,因此阐明这些通讯关系至关重要。
CellPhoneDB配有详实的受配体数据库,其整合了此前的公共数据库,还会手动矫正,以得到更加准确的受配体注释。此外,针对受配体有多个亚基的情况,也进行了注释。下面这张图显示了CellPhoneDB配有的数据库包含多少种分泌蛋白和膜蛋白、蛋白质复合物、受配体关系,以及它们来源于什么数据库。
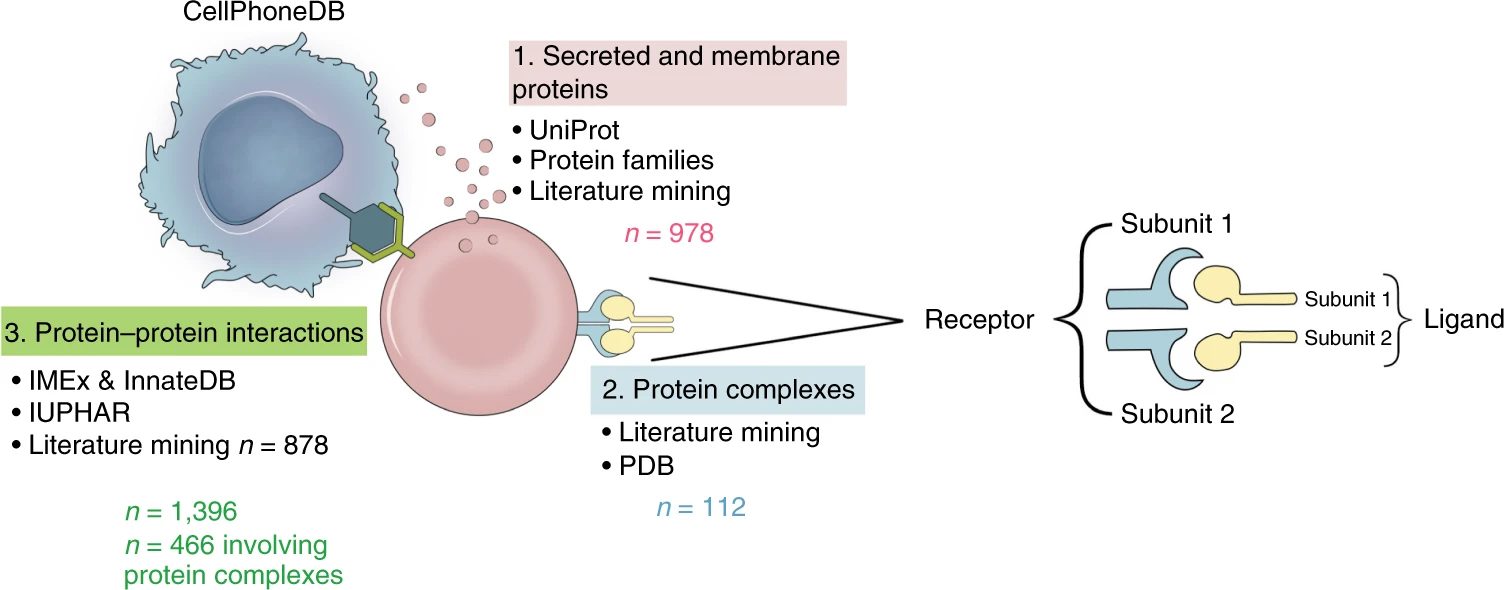
1. CellPhoneDB推断细胞通讯的原理
在给定表达矩阵和细胞注释之后,对于gene1-gene2这个互作关系,计算某一个clusterA里面gene1的表达均值,计算另一个clusterB中gene2的表达均值,二者的均值为MEAN;在随机更换细胞的label之后,依据新的标签,计算“clusterA”里面gene1的表达均值,"clusterB"中gene2的表达均值,再求一个平均值mean,这样的过程重复多次,就可以得到一个mean的分布,即null distribution。MEAN在这个分布中所在的位置以及更极端的位置,构成的占比,就是p值(p值的定义)。所以CellPhoneDB推测两种细胞类型之间显著富集的受配体关系,本质上还是基于一个细胞类型里面的受体表达量,以及另一种细胞类型里面的配体表达量。此外,如果某种关系无处不在(在所有细胞类型之间都很明显),则找不出来。
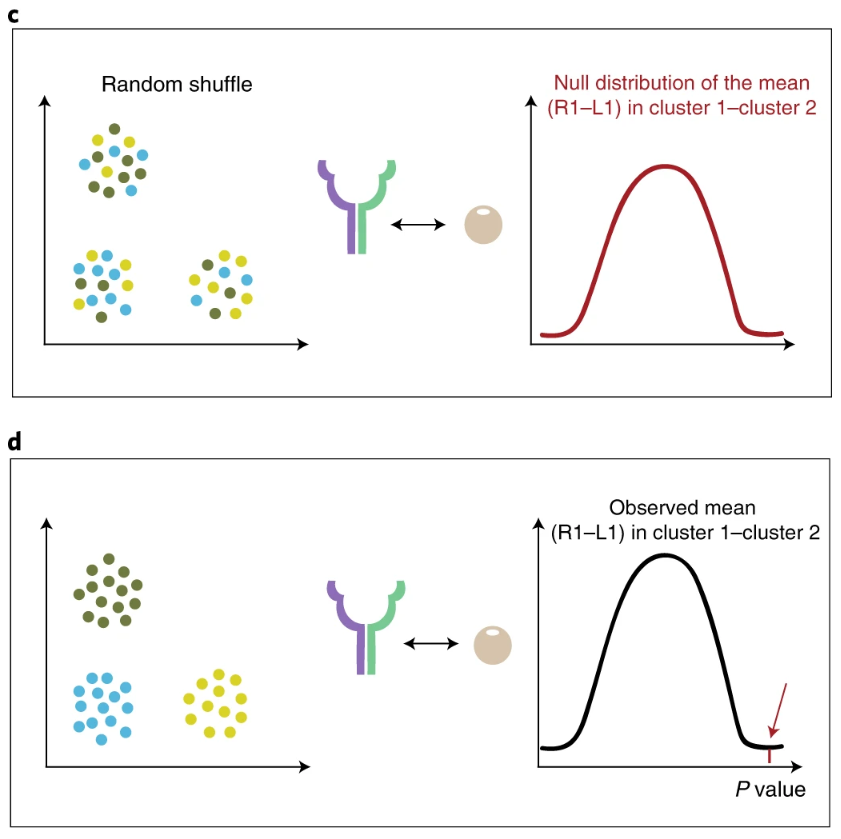
此外还有几个需要注意的地方:
- 大样本时会下采样,只分析1/3的细胞
- 多个亚基时考虑表达低的那一个亚基
- 表达占比达到一定阈值的基因才会被分析,默认是10%
2. 如何展示结果
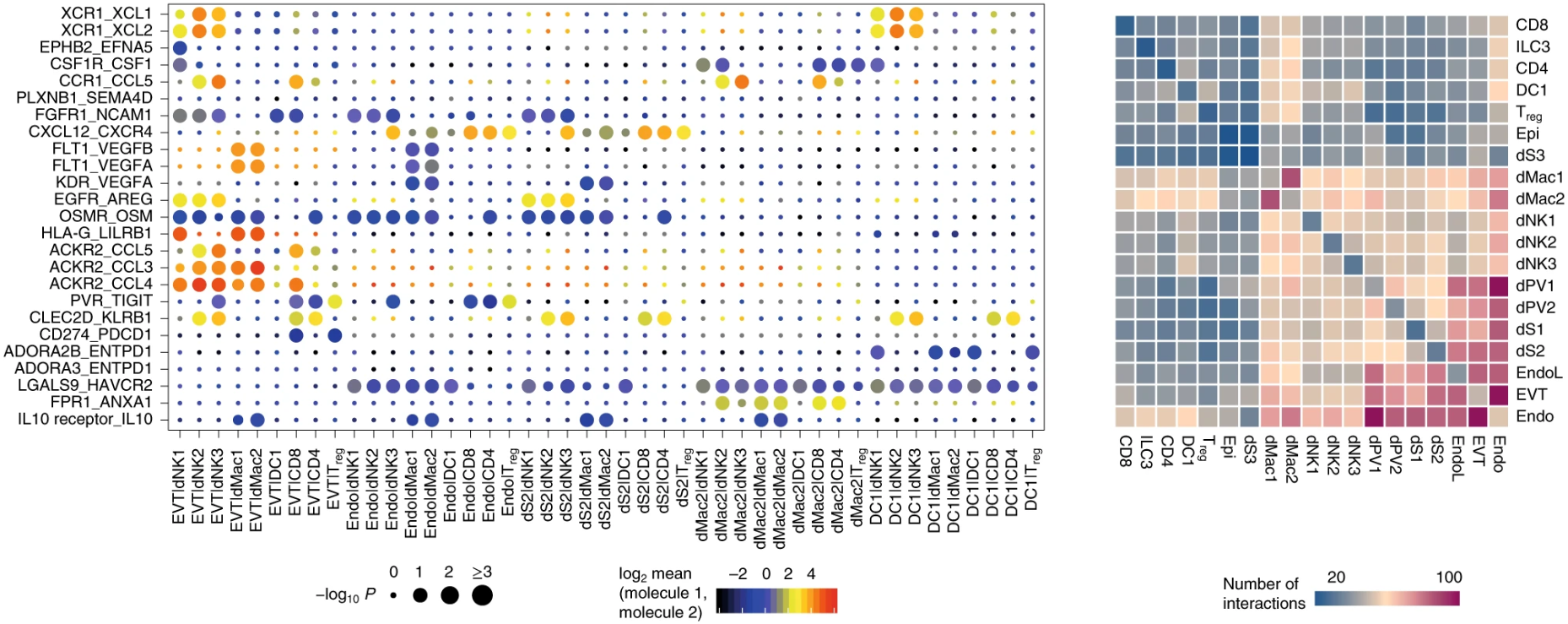
这是原文献给的可视化例子,这里有两个地方需要注意:
- 右边的热图表示细胞类型两两之间的相互作用的数量,我们可以看到沿着对角线,左右是对称的,也就是A-B与B-A的互作数目是一样的,为什么会这样?
- 左边是具体受配体对,细胞对的互作气泡图,点的大小表示显著水平,颜色则是The means of the average expression level of interacting molecule 1 in cluster 1 and interacting molecule 2 in cluster 2 注意到了吗,说的是interacting molecule 1/2,而没有说哪一个是受体哪一个是配体。
原因都和CellPhoneDB内置的gene-gene互作关系列表有关。CellPhoneDB区分不了受体还是配体,对于gene1-gene2,可以是gene1配体gene2受体,也可以是gene1受体gene2配体(如下图)。我个人觉得也是由于这个原因,右边那个热图为了说起来方便,才把不管做受体还是做配体的关系都算作是两种细胞的互作关系,因此A-B和B-A在热图中的数值是一样的(不然横纵坐标写个interacting molecule,看到的人自然会问,这个分子是受体还是配体呢,加一起就省事了——都包含)。
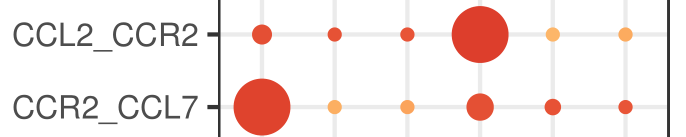
这一点,github有提到:
也是这个原因,我看到文章如果用了CellPhoneDB的话,会留意它的图,如果是用有向图表示细胞群两两之间的关系数量,我会想这样做合不合适(当然是不合适的)
3. 实际分析
公众号后台回复
20210723获取本次演示的测试数据,以及主要的可视化代码。
3.1 输入文件的格式
注释文件
一共两列,Cell列cell_type列,有列名;.csv, .txt后缀都行
表达文件
normalize之后的矩阵,一般简单相除normalize一下就行;.csv, .txt后缀都行
3.2 运行
软件的安装这里就不讲了,创建一个conda环境,pip install下载安装就可以了
运行CellPhoneDB的主代码很简单:
source /home/huangsiyuan/miniconda3/bin/activate cpdb
file_count=/home/huangsiyuan/cpdb/test_normat.txt
file_anno=/home/huangsiyuan/cpdb/test_anno.txt
outdir=/home/huangsiyuan/cpdb/test
if [ ! -d ${outdir} ]; then
mkdir ${outdir}
fi
cellphonedb method statistical_analysis \
--counts-data hgnc_symbol \
--output-path ${outdir} \
--threshold 0.01 \ #Percentage of cells expressing the specific ligand or receptor
--threads 10 \
${file_anno} ${file_count}
source /home/huangsiyuan/miniconda3/bin/deactivate cpdb
#如果细胞数太多,可以添加下采样参数,默认只分析1/3的细胞
#--subsampling
#--subsampling-log true #对于没有log转化的数据,还要加这个参数
这一步之后在test文件夹里面会生成4个文件
deconvoluted.txt
means.txt
pvalues.txt
significant_means.txt
其中,
means.txt行是受配体pair,列是细胞pair,值为受体、配体在相应的cluster中表达均值的平均数;pvalues.txt格式与means.txt类似,值为p值;significant_means.txt格式和内容都与means.txt类似,不过仅保留了p值小于0.05的平均数。
4. 结果的可视化
在这一步中,我一般只用到上述的means.txt和pvalues.txt文件
我们还是先仿照文献原文,画出那两张图
library(tidyverse)
library(RColorBrewer)
library(scales)
pvalues=read.table("./test/pvalues.txt",header = T,sep = "\t",stringsAsFactors = F)
pvalues=pvalues[,12:dim(pvalues)[2]] #此时不关注前11列
statdf=as.data.frame(colSums(pvalues < 0.05)) #统计在某一种细胞pair的情况之下,显著的受配体pair的数目;阈值可以自己选
colnames(statdf)=c("number")
#排在前面的分子定义为indexa;排在后面的分子定义为indexb
statdf$indexb=str_replace(rownames(statdf),"^.*\\.","")
statdf$indexa=str_replace(rownames(statdf),"\\..*$","")
#设置合适的细胞类型的顺序
rankname=sort(unique(statdf$indexa))
#转成因子类型,画图时,图形将按照预先设置的顺序排列
statdf$indexa=factor(statdf$indexa,levels = rankname)
statdf$indexb=factor(statdf$indexb,levels = rankname)
statdf%>%ggplot(aes(x=indexa,y=indexb,fill=number))+geom_tile(color="white")+
scale_fill_gradientn(colours = c("#4393C3","#ffdbba","#B2182B"),limits=c(0,20))+
scale_x_discrete("cluster 1 produces molecule 1")+
scale_y_discrete("cluster 2 produces molecule 2")+
theme_minimal()+
theme(
axis.text.x.bottom = element_text(hjust = 1, vjust = NULL, angle = 45),
panel.grid = element_blank()
)
ggsave(filename = "interaction.num.1.pdf",device = "pdf",width = 12,height = 10,units = c("cm"))
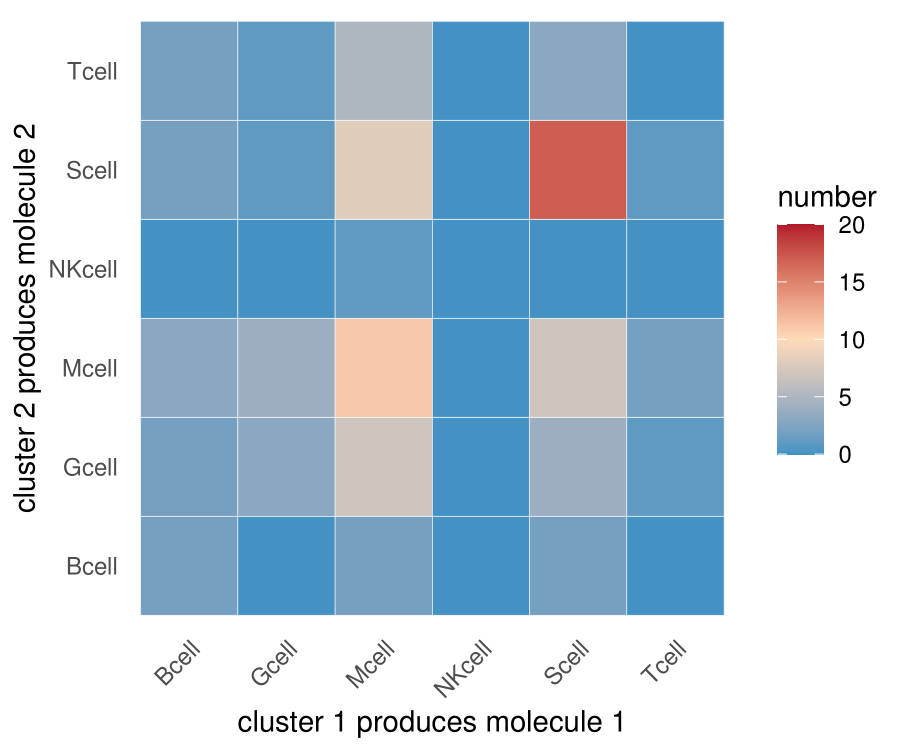
这里与文献中图不一致的地方是,我这个图并不是关于对角线对称的,因为我没有将A-B,B-A的互作关系求和
举个例子
在CellPhoneDB输出的结果中,经统计,A-B有10个显著的互作关系,B-A有20个显著的互作关系【①】。然而A-B的互作其实包含A做配体8次,A做受体2次,B-A的互作其实包含B做配体19次,B做受体1次,所以严格来讲,A和B两种细胞互作,A做配体9次,B做配体21次【②】,这些信息是CellPhoneDB给不了的。当然互作关系还是共计30次【③】。
换言之,文献中对称的图给的信息③,我上面那个图给的信息①,信息②是不知道的(如果肉眼一个一个去看CellPhoneDB数据库中gene1-gene2哪个是受体哪个是配体,还是可以统计出来的)。
因本文篇幅较长,余下的可视化部分将在下一篇展示,敬请期待~
参考文献
[1] Efremova M, Vento-Tormo M, Teichmann S A, et al. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes[J]. Nature protocols, 2020, 15(4): 1484-1506.
因水平有限,有错误的地方,欢迎批评指正!
单细胞分析实录(18): 基于CellPhoneDB的细胞通讯分析及可视化 (上篇)的更多相关文章
- 单细胞分析实录(19): 基于CellPhoneDB的细胞通讯分析及可视化 (下篇)
在上一篇帖子中,我介绍了CellPhoneDB的原理.实际操作,以及一些值得注意的地方.这一篇继续细胞通讯分析的可视化. 公众号后台回复20210723获取本次演示的测试数据,以及主要的可视化代码. ...
- (二): 基于ZeroMQ的实时通讯平台
基于ZeroMQ的实时通讯平台 上篇:C++分布式实时应用框架 (Cpp Distributed Real-time Application Framework)----(一):整体介绍 通讯平台作为 ...
- 【代码更新】单细胞分析实录(20): 将多个样本的CNV定位到染色体臂,并画热图
之前写过三篇和CNV相关的帖子,如果你做肿瘤单细胞转录组,大概率看过: 单细胞分析实录(11): inferCNV的基本用法 单细胞分析实录(12): 如何推断肿瘤细胞 单细胞分析实录(13): in ...
- 单细胞分析实录(8): 展示marker基因的4种图形(一)
今天的内容讲讲单细胞文章中经常出现的展示细胞marker的图:tsne/umap图.热图.堆叠小提琴图.气泡图,每个图我都会用两种方法绘制. 使用的数据来自文献:Single-cell transcr ...
- 【代码更新】单细胞分析实录(21): 非负矩阵分解(NMF)的R代码实现,只需两步,啥图都有
1. 起因 之前的代码(单细胞分析实录(17): 非负矩阵分解(NMF)代码演示)没有涉及到python语法,只有4个python命令行,就跟Linux下面的ls grep一样的.然鹅,有几个小伙伴不 ...
- 高性能Linux服务器 第10章 基于Linux服务器的性能分析与优化
高性能Linux服务器 第10章 基于Linux服务器的性能分析与优化 作为一名Linux系统管理员,最主要的工作是优化系统配置,使应用在系统上以最优的状态运行.但硬件问题.软件问题.网络环境等 ...
- 【GWAS文献】基于GWAS与群体进化分析挖掘大豆相关基因
Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improv ...
- 基于Keil C的覆盖分析,总结出编程中可能出现的几种不可预知的BUG
基于Keil C的覆盖分析,总结出编程中可能出现的几种不可预知的BUG,供各位网友参考 1.编译时出现递归警告,我看到很多网友都采用再入属性解决,对于再入函数,Keil C不对它进行覆盖分析,采用模拟 ...
- 基于Petri网的工作流分析和移植
基于Petri网的工作流分析和移植 一.前言 在实际应用场景,包括PEC的订单流程从下订单到订单派送一直到订单完成都是按照一系列预先规定好的工作流策略进行的. 通常情况下如果是采用面向过程的编程方法, ...
随机推荐
- 2019个嵌入式市场研究,持续的C/C++优势
2019个嵌入式市场研究,持续的C/C++优势 2019 Embedded Markets Study reflects emerging technologies, continued C/C++ ...
- C#后台定义一个DataTable并手动写入静态数据(测试数据)
//创建一个DataTable,并为之添加数据(自定义DataTable) DataTable dtz = new DataTable(); //添加Table中的列 DataColumn dc1 = ...
- Mac设置charles证书信任
- UF_CSYS 坐标系操作
Open C UF_CSYS_ask_csys_info 获取WCS坐标系的原点坐标和矩阵标识UF_CSYS_ask_matrix_of_object 获得对象 ...
- Java第二次博客作业
Java第二次博客作业 时间过的很快啊,在不知不觉中这门课程的学习也就快要过去一半了,现在就来总结一下在这个第二个月的学习当中存在的问题以及得到的心得. 1.前言 第四次题目集和第五次题目集给我的感觉 ...
- Zookeeper 分布式锁 (图解+秒懂+史上最全)
文章很长,而且持续更新,建议收藏起来,慢慢读! 高并发 发烧友社群:疯狂创客圈(总入口) 奉上以下珍贵的学习资源: 疯狂创客圈 经典图书 : 极致经典 + 社群大片好评 < Java 高并发 三 ...
- git stash回退
目录 1.起因 2. 问题出现 3.修复 4. 注意 1.起因 这个问题要从今天刚遇到的事儿说起,昨晚代码出了个乌龙事件,本来正在dev分支进行新功能的开发,但是测试出现的问题是在release 分支 ...
- Arduino参考手册-函数和变量及电路图
标题: Arduino参考手册-函数和变量及电路图 作者: 梦幻之心星 sky-seeker@qq.com 标签: [#Arduino,#参考手册,#函数,#变量] 目录: [Arduino] 日期: ...
- 手把手教你IDEA连接码云(Gitee)
目录 前言 一.下载.安装git 1.打开git官网,选择你的操作系统 2.根据你的系统位数选择相应的版本下载 3.安装 4.配置全局的用户名.邮箱 5.在idea中配置git目录 二.配置Gitee ...
- testt
一级标题 二级标题 三级标题 四级标题 l 1