miRAN 分析以及mRNA分析
一些参考资料
http://www.360doc.com/content/17/0528/22/19913717_658086490.shtml
https://www.cnblogs.com/triple-y/p/9338890.html
一、对miRNA进行分析
1、bowtie比对
"bowtie -q -v 2 -l 10 -k 15 /data/pub/shehb/Spinach_genome/spinach_genome_v1.fa "+fq+" -S "+fq+".sam >mapping.info"
2、HTSeq 计算reads数量 (详细:http://www.chenlianfu.com/?p=2438)
(1)HTSeq是对有参考基因组的转录组测序数据进行表达量分析的,其输入文件必须有SAM和GTF文件。
(2)一般情况下HTSeq得到的Counts结果会用于下一步不同样品间的基因表达量差异分析,而不是一个样品内部基因的表达量比较。因此,HTSeq设置了-a参数的默认值10,来忽略掉比对到多个位置的reads信息,其结果有利于后续的差异分析。
(3)输入的GTF文件中不能包含可变剪接信息,否则HTSeq会认为每个可变剪接都是单独的基因,导致能比对到多个可变剪接转录本上的reads的计算结果是ambiguous,从而不能计算到基因的count中。即使设置-i参数的值为transcript_id,其结果一样是不准确的,只是得到transcripts的表达量。
run = "htseq-count -s no -t mRNA -i ID "+sam+" ./Spinach_genome/spinach_gene_v1miRNA.gff3 > "+sam+"count_out.txt"
参数:
-f | --format default: sam 设置输入文件的格式,该参数的值可以是sam或bam
-r | --order default: name 设置sam或bam文件的排序方式,该参数的值可以是name或pos。,但HTSeq推荐使用name排序,且一般比对软件的默认输出结果也是按name进行排序的。
-s | --stranded default: yes 设置是否是链特异性测序。该参数的值可以是yes,no或reverse。no表示非链特异性测序;若是单端测序,yes表示read比对到了基因的正义链上;若是双末端测序,yes表示read1比对到了基因正义链上,read2比对到基因负义链上;reverse表示双末端测序情况下与yes值相反的结果。根据说明文件的理解,一般选择no, yes是成对的reads数量-a | --a default: 10 忽略比对质量低于此值的比对结果。在0.5.4版本以前该参数默认值是0。 -t | --type default: exon 程序会对该指定的feature(gtf/gff文件第三列)进行表达量计算,而gtf/gff文件中其它的feature都会被忽略。 比如:在mRNA中,选择exon,表示只计算exon的reads数量-i | --idattr default: gene_id 设置feature ID是由gtf/gff文件第9列那个标签决定的;若gtf/gff文件多行具有相同的feature ID,则它们来自同一个feature,程序会计算这些features的表达量之和赋给相应的feature ID。 看基因前面的标志是什么-m | --mode default: union 设置表达量计算模式。该参数的值可以有union, intersection-strict and intersection-nonempty。这三种模式的选择请见上面对这3种模式的示意图。从图中可知,对于原核生物,推荐使用intersection-strict模式;对于真核生物,推荐使用union模式。 -o | --samout 输出一个sam文件,该sam文件的比对结果中多了一个XF标签,表示该read比对到了某个feature上。 -q | --quiet 不输出程序运行的状态信息和警告信息。 -h | --help 输出帮助信息。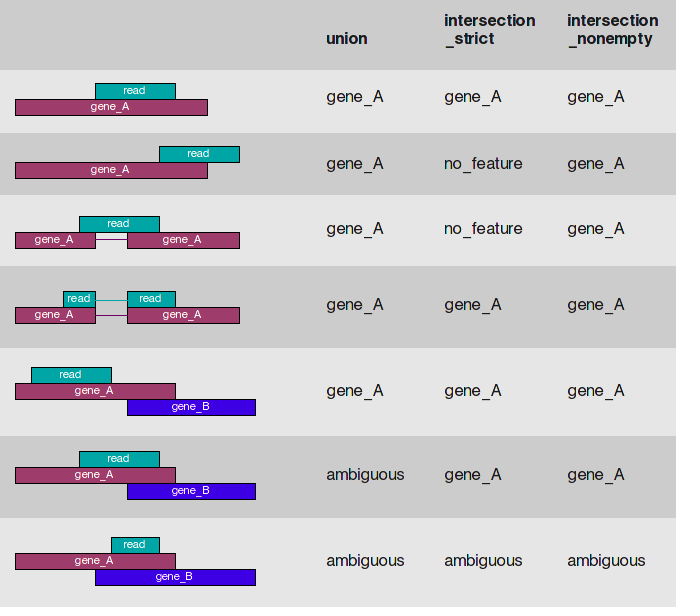
1 paste *.txt |awk '{printf $1"\t";for(i=2;i<=18;i+=2)printf $i"\t";printf $i"\n"}' |less -S
2
3 ##18 为总共有多少列
3、对于有重复的样本用DESeq2进行分析
自认为感觉不是很好安装包 ,我用下前面方法安装上去的
若R安装包出现package ‘xx’ is not available (for R version 3.4.2)的问题时:
用以下方法安装包
source("http://bioconductor.org/biocLite.R")
biocLite("包名")
DESeq2对于输入数据的要求
1.DEseq2要求输入数据是由整数组成的矩阵。
2.DESeq2要求矩阵是没有标准化的。
DESeq2进行差异表达分析
DESeq2包分析差异表达基因简单来说只有三步:构建dds矩阵,标准化,以及进行差异分析。
1、构建dds矩阵
需要:(1)countData, 是readscount计算后,合并的形成的矩阵。行为基因名字,列为样本
(2)colData, 样本信息,标出哪些是control,哪些是treat。第一列样品名称(其实就是countData第一行的名称),第二列是样品处理情况(对照还是处理等),condition的类型是一个factor。
(3)差异比较矩阵 即上述代码中的design。 差异比较矩阵就是告诉差异分析函数是要从要分析哪些变量间的差异,简单说就是说明哪些是对照哪些是处理。
library(S4Vectors)
library(stats4)
library(BiocGenerics)
library(parallel)
library(IRanges)
library(GenomicRanges)
library(GenomeInfoDb)
library(SummarizedExperiment)
library(Biobase)
library(DelayedArray)
library(matrixStats)
library(DelayedArray)
library(DESeq2)
library(ggplot2)
setwd("/Users/hongbingshe/Desktop")
condition <- factor(c("XX","XX","XX","XY","XY","XY"))
countData <- read.table("XX_XYmiRNA.count_out",header = T,sep = "\t",row.names = "X") ##open file
colData <- data.frame(row.names = colnames(countData),condition)
dds <- DESeqDataSetFromMatrix(countData,DataFrame(condition),design = ~ condition)
head(dds) ##
dds2 <- DESeq(dds) ###normalize rlogTransformation(dds2). ???
resultsNames(dds2) ###objective name
res <- results(dds2)
summary(res) ###infomation
table(res$padj<0.05) ##number of true 小于0.05 的基因个数
res <- res[order(res$padj),]
diff_gene_deseq2 <- subset(res,padj <0.05 & (log2FoldChange >1 |log2FoldChange < -1)) ###FC >1, P <0.05
differ_gene_deseq2 <- row.names(diff_gene_deseq2)
resdata <- merge(as.data.frame(res),as.data.frame(counts(dds2,normalize=TRUE)),by="row.names",sort=FALSE)
write.csv(resdata,file = "/Users/hongbingshe/Desktop/XX_XYmiRNA.cvs",row.names = F) ##output file
resdata[which(resdata$padj < 0.05 & resdata$log2FoldChange > 1 ),'significant'] <-'up'
resdata[which(resdata$padj < 0.05 & resdata$log2FoldChange < -1 ),'significant'] <-'down'
resdata[!resdata$significant%in%c('up','down'),'significant'] <- 'no'
p <- ggplot(resdata,aes(resdata$log2FoldChange,-log10(resdata$padj))) + geom_point(aes(color=significant),size=0.5) +scale_color_manual(limits=c('up','down','no'),values = c('blue','red','gray40')) +labs(x="log2(Fold Change)",y="-log10(P-value)")
p <- p + theme(panel.grid.major = element_line(color="gray",size=0.2), panel.background = element_rect(color = "black",fill = "transparent")) +geom_vline(xintercept = c(-1,1),color="gray",linetype=2,size=0.5)+geom_hline(yintercept = -log10(1),color='gray',linetype=2,size=0.5)
p <- p + theme(legend.title = element_blank(),legend.key = element_rect(fill='transparent'),legend.background = element_rect(fill = 'transparent'))
p <- p + xlim(-4,4) +ylim(0,40) ##X scale
p
ggsave("XY_XXmiRNAvalcano_plot.png",p,width = 4.5,height=5.5)
标记出你期望的基因名字
(1)首先需要做一个数据框. 假如为test
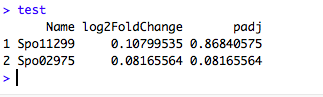
(2)导入 ggrepel 包
library(ggrepel)
(3)添加图层
p + geom_text_repel(data =test,aes(test$log2FoldChange,-log10(test$padj),label=test$Name))
或者用p +geom_label_repel(data = test,aes(test$log2FoldChange,-log10(test$padj),label=test$Name))
二、mRNA分析
选用hisat2+HTseq+DEseq2
1、hisat2:
run = "hisat2 -x ./Spinach_genome/spinach_genome_v1.fa -p 4 -1 "+i+" -2 "+j+" -S "+Out
2、HTSeq2
htseq-count -f bam -r name -s no -a 10 -t exon -i ID -m intersection-nonempty yourfile_name.bam ~/reference/hisat2_reference/Homo_sapiens.GRCh38.86.chr_patch_hapl_scaff.gtf > counts.txt
3、DEseq2
同上
关注下方公众号可获得更多精彩

miRAN 分析以及mRNA分析的更多相关文章
- 常用 Java 静态代码分析工具的分析与比较
常用 Java 静态代码分析工具的分析与比较 简介: 本文首先介绍了静态代码分析的基 本概念及主要技术,随后分别介绍了现有 4 种主流 Java 静态代码分析工具 (Checkstyle,FindBu ...
- Memcached源代码分析 - Memcached源代码分析之消息回应(3)
文章列表: <Memcached源代码分析 - Memcached源代码分析之基于Libevent的网络模型(1)> <Memcached源代码分析 - Memcached源代码分析 ...
- mysql 分析3使用分析sql 性能 show profiles ;
show variables like '%profiling%'; 查看状态 查看时间去哪了``` set profiling=1;// 打开 show profiles; 查看执行过的 ...
- [转载] 常用 Java 静态代码分析工具的分析与比较
转载自http://www.oschina.net/question/129540_23043 简介: 本文首先介绍了静态代码分析的基本概念及主要技术,随后分别介绍了现有 4 种主流 Java 静态代 ...
- x264源代码简单分析:宏块分析(Analysis)部分-帧间宏块(Inter)
===================================================== H.264源代码分析文章列表: [编码 - x264] x264源代码简单分析:概述 x26 ...
- x264源代码简单分析:宏块分析(Analysis)部分-帧内宏块(Intra)
===================================================== H.264源代码分析文章列表: [编码 - x264] x264源代码简单分析:概述 x26 ...
- loadrunner 结果分析-loadrunner结果分析
结果分析-loadrunner结果分析 by:授客 QQ:1033553122 百度网盘分享链接: 烦请 复制一下网址到浏览器中打开,输入密码提取 链接: http://pan.baidu.com/s ...
- ⑥NuPlayer播放源码分析之DecoderBase分析
NuPlayer播放源码分析之DecoderBase分析 [时间:2017-02] [状态:Open] [关键词:android,nuplayer,开源播放器,播放框架,DecoderBase,Med ...
- 【转载】常用 Java 静态代码分析工具的分析与比较
摘自:http://www.oschina.net/question/129540_23043常用 Java 静态代码分析工具的分析与比较 简介: 本文首先介绍了静态代码分析的基本概念及主要技术,随后 ...
随机推荐
- MySQL:提高笔记-5
MySQL:提高笔记-5 学完基础的语法后,进一步对 MySQL 进行学习,前几篇为: MySQL:提高笔记-1 MySQL:提高笔记-2 MySQL:提高笔记-3 MySQL:提高笔记-4 MySQ ...
- the Agiles Scrum Meeting 博客汇总
the Agiles 团队博客目录 一.Scrum Meeting 1. Alpha the Agiles Scrum Meeting 1 the Agiles Scrum Meeting 2 the ...
- BUAA_2020_软件工程_热身作业
项目 内容 这个作业属于哪个课程 2020春季计算机学院软件工程(罗杰 任建) 这个作业的要求在哪里 热身作业要求 我在这个课程的目标 了解软件工程的技术,掌握工程化开发的能力 这个作业在哪个具体方面 ...
- rabbitmq生产者消息确认
在使用 RabbitMQ 的时候,有时候当我们生产者发送一条消息到 RabbitMQ 服务器后,我们 生产者想知道消息是否到达了 RabbitMQ 服务器上.这个时候我们应该如何处理? 针对上述问题, ...
- FastAPI 学习之路(五十六)将token存放在redis
在之前的文章中,FastAPI 学习之路(二十九)使用(哈希)密码和 JWT Bearer 令牌的 OAuth2,FastAPI 学习之路(二十八)使用密码和 Bearer 的简单 OAuth2,Fa ...
- TVS管性能及选型总结
https://wenku.baidu.com/view/5b5bda5526fff705cc170af8.html
- 计算机网络之网络层路由选择协议(自治系统AS、RIP、OSPF、BGP)
文章转自:https://blog.csdn.net/weixin_43914604/article/details/105313629 学习课程:<2019王道考研计算机网络> 学习目的 ...
- IDA*、操作打表、并行处理-The Rotation Game HDU - 1667
万恶之源 优秀题解 用文字终究难以穷尽代码的思想 思路 每次操作都有八种选择,相当于一棵每次延申八个子节点的搜索树,故搜索应该是一种方法.而这题要求求最少步数,我们就可以想到可以试试迭代加深搜索(但其 ...
- (类)Program1.1
1 class MyClass: 2 3 i = 12345 4 5 def __init__(self): 6 self.data = "WOOWOWOWO" 7 8 def f ...
- hdu 1158 Employment Planning(DP)
题意: 有一个工程需要N个月才能完成.(n<=12) 给出雇佣一个工人的费用.每个工人每个月的工资.解雇一个工人的费用. 然后给出N个月所需的最少工人人数. 问完成这个项目最少需要花多少钱. 思 ...