群体遗传之ped格式
1、PED简介
PED文件格式是广泛使用的用于连锁系谱数据分析的格式,并用作plink程序的输入。PLINK是一个免费的,开源的全基因组关联分析工集,旨在以高计算效率的方式执行一系列基本的,大规模的分析。PED能够处理二倍体SNP数据。
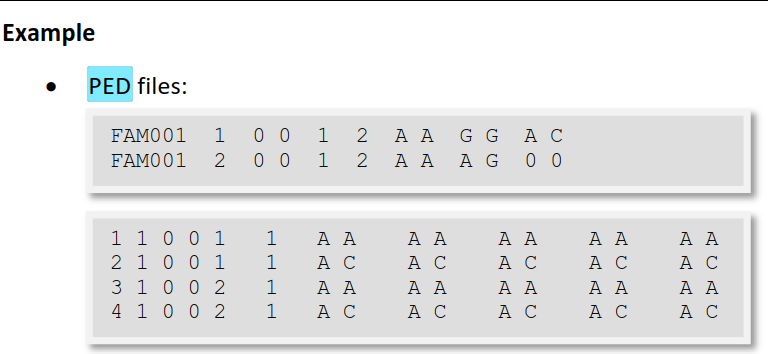
空格(空格或制表符)分隔的文本文件*.ped
每一行对应一个individual
以下前6列是必须的(id是字母数字):
o Family ID (Family ID用来表示家族,同一个家族用同一个family ID表示)
o Individual ID (用来表示个体,family ID和Individual ID连起来必须能够唯一表示每个样本)
o Paternal ID (表示父本ID,)
o Maternal ID (母本ID,)
o Sex (1代表male,2代表female, 其他数字表示unknown。)
o Phenotype (代表表型,其中表型可以是离散型的(比如关联性状),也可以是连续型的(比如数量性状),plink会自动识别对应的类型。通过以上6个必须的字段,可以完整的映射到某一性状的家系图上。)
关联性状应该这样编码:
o -9 missing
o 0 missing
o 1 unaffected
o 2 affected
column 7 onwards: Genotypes (对于关联分析而言,除了表型相关信息,还需要基因型信息)
any character (e.g.: 1,2,3,4 or A,C,G,T or anything else)
missing genotype: 0
所有的标记必须是双等位的(二倍体)。要么两个等位基因都缺失,要么两者都不缺失。单倍体数据:编码为二倍体纯合子。两个等位基因依次出现。
Comments: line starts with #
在ped文件中,每个snp位点的基因型需要两列来表示,分别表示major allel 和 minor allel。在表示基因型时,既可以使用A,C,G,T字母的形式,也可以采用1,2数字编码的形式。默认情况下,用0来表示基因型的缺失。
2、MAP简介
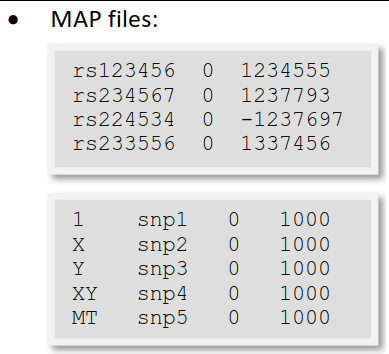
MAP文件的每一行描述一个 single marker且必须包含4列:
chromosome (1-22, X, Y, MT or 0 if unplaced) #染色体编号为数字, 未知为0
rs# or snp identifier #SNP名称为字符或数字, 可以从1编号, 注意要和bed文件SNP列一一对应
Genetic distance (morgans) (missing: 0) #遗传距离(摩尔)
SNP物理坐标
MAP必须包含与PED文件中一样多的markers。‘
PED文件中的标记不需要按照基因组顺序排列,但是MAP应该与PED文件maker顺序一致
PGDSpider软件中对于不同的文件格式有一个详细的说明。
http://pngu.mgh.harvard.edu/~purcell/plink/data.shtml#ped
群体遗传之ped格式的更多相关文章
- plink:ped格式转换为bed格式
命令行如下: plink --file FILENAME --make-bed --out FILENAME 第一个FILENAME的后缀为.ped和.map,生成的第二个FILENAME的后缀为.b ...
- 重测序(RADseq)做群体遗传分析套路
实验材料 构建的群体,或自然群体,如各地方品种. RAD文库构建 提取DNA后,构建文库,简要步骤如下: ① 限制性内切酶TaqI酶切: ② 连接P1接头: ③ DNA随机打断片断化: ④ 目的片段回 ...
- 【转】群体研究套路:开心果denovo+重测序+转录组+群体进化+选择位点
转自公众号Eric生信小班.学习群体遗传套路 中科院昆明动物园吴东东研究团队联合国外研究团队2019年在Genome Biology发表题为Whole genomes and transcriptom ...
- 用popart构建常染色体单倍型网络(Autosomal haplotypes network construction with popart)
1)将vcf转化为plink格式,假定输入的vcf文件名为:17893893-17898893.vcf,也可以参考链接:将vcf文件转化为plink格式并且保持phasing状态 /vcftools ...
- Eigensoft-smartpca分析PCA报错:warning (mapfile): bad chrom: Segmentation fault
目录 问题 解决 问题 一直以来用Eigensoft的smartpca来做群体遗传的PCA分析很顺畅,结果也比较靠谱. 但今天报错如下: $ ~/miniconda3/bin/smartpca -p ...
- 千人基因组计划数据库下载某段区域SNP
进入http://browser.1000genomes.org/index.html网站 假定要寻找“6:133098746-133108745”这段距离的SNP数据,“6”表示6号染色体,后面的数 ...
- 遗传算法详解(LINGO及MatlabGA工具箱求解实现)
遗传算法 1.前言 遗传算法是一种基于生物界自然群体遗传进化机制的自适应全局优化概率搜索算法.它与传统算法不同,不依赖梯度信息,而是通过模拟自然进化过程来搜索最优解. 例子:兔子的遗传进化 有人说,现 ...
- GWAS:拒绝假阳性之case和control数量比例严重失衡的解决方案(SAIGE模型的应用)
一.为什么要校正case和control数量比例不平衡情况 试问作为生信届人员,最怕的是什么,当然是统计结果不靠谱.统计结果不靠谱包括两方面:一个是假阴性,一个是假阳性.假阴性可以理解为白天鹅被误当成 ...
- Python的浮点数损失精度问题
本篇讨论的现象可以从下面这段脚本体现出来: >>> x = 0.0 >>> for i in range(10): x += 0.1 print(x) 0.1 0. ...
随机推荐
- C++ 标准库,可变参数数量,参数类型相同
#include <iostream> // 可变模板参数 // 此例:可以构造可变数量,可变类型的函数输入. // 摘自:https://www.cnblogs.com/qicosmos ...
- C# List<T> 转 DataTable
C# List<T>转DataTable 学习自:博客园 Overview 数据!!个人认为程序就是将数据变着花样的显示它.那么这个时候我们的数据处理和获取就时我们的关键一步,如果你数据都 ...
- Codeforces Round #603 (Div. 2) E. Editor 线段树
E. Editor The development of a text editor is a hard problem. You need to implement an extra module ...
- js a 标签 通过download 实现下载功能
download 属性规定被下载的超链接目标. 在 <a> 标签中必须设置 href 属性. 该属性也可以设置一个值来规定下载文件的名称.所允许的值没有限制,浏览器将自动检测正确的文件扩展 ...
- MySQL属性SQL_MODE学习笔记
最近在学习<MySQL技术内幕:SQL编程>并做了笔记,本博客是一篇笔记类型博客,分享出来,方便自己以后复习,也可以帮助其他人 SQL_MODE:MySQL特有的一个属性,用途很广,可以通 ...
- java循环定时器@Scheduled的使用
@Scheduled 注解 用于定时循环执行任务 例如: @Scheduled(cron="0 */10 * * * ?") 表示每隔十分钟执行一次 每隔5秒执行一次:" ...
- SonarLint各种提示的意思
1.Refactor this method to reduce its Cognitive Complexity from 29 to the 15 allowed. 2.Method has 15 ...
- IDEA maven设置配置
IDEA Maven配置 1. 下载maven 下载地址 从官网上,下载一个压缩包,然后解压到任意的文件夹 Maven的安装必须需要jdk1.7+ 2. 环境变量设置 M2_HOME改为具体的路径,其 ...
- c# winform 窗体失去焦点关闭(钩子实现)
先来一个辅助类 using System; using System.Collections.Generic; using System.Linq; using System.Runtime.Inte ...
- python基础(5):格式化输出、基本运算符、编码问题
1. 格式化输出 现在有以下需求,让⽤户输入name, age, job,hobby 然后输出如下所⽰: ------------ info of Alex Li ----------- Name : ...