Uncovering thousands of new peptides with sequence-mask-search hybrid de novo peptide sequencing framework (使用序列掩码搜索结合肽段从头测序框架发现了数千个新肽段)-解读人:刘佳维
期刊名:Molecular & Cellular Proteomics
发表时间:(2019年12月)
IF:4.828
单位:
- 朱拉隆功大学
- 费城威斯塔研究所
物种:人
技术:de novo从头测序,深度学习
一、 概述:
该研究开发了一种基于深度学习的肽段从头测序框架SMSNet,在保持良好的识别覆盖率的同时,氨基酸准确度能达到95%以上。SMSNet揭示了超过10000个以前未分类的人类白细胞抗原(HLA)和磷酸肽,并结合数据库搜索方法,将肽鉴定的覆盖范围扩大了近30%。
二、 研究背景:
基于质谱的蛋白质组学数据的典型分析方法数据库搜索方法仅识别在参考数据库中存在的氨基酸序列,限制了发现新的肽的可能性,例如那些含有未经特征化突变的肽,或者那些起源于rna和蛋白质的意外加工的肽。相反,从头测序方法直接从观察到的质谱中确定氨基酸序列,但有时精度较低。尽管近年来已经证明了深度学习可以有效地应用于从头测序问题,但从头测序方法和数据库搜索方法在识别肽的准确性和数量方面仍存在巨大的性能差距。这一限制的关键部分在于质谱谱图是有噪声的,有时会缺乏关键信息。如何解决这一问题是提升从头测序方法的关键所在。
三、实验设计:

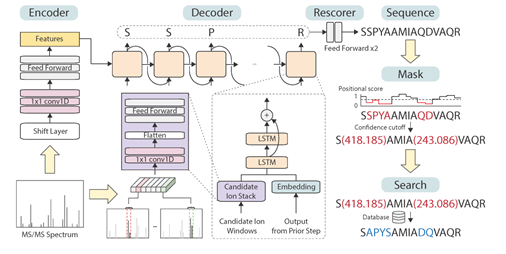
SMSNet序列掩码搜索框架
四、研究成果:
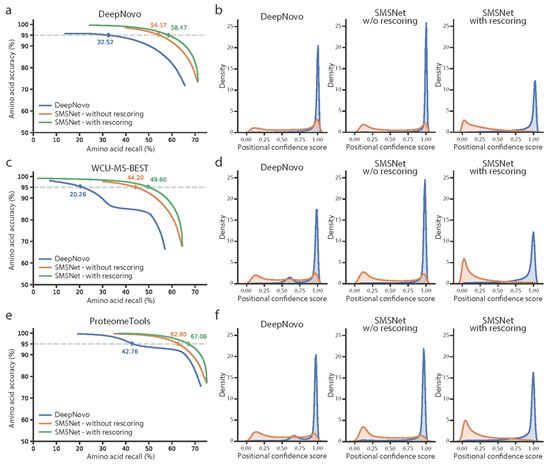
1、与目前最新的de novo测序软件DeepNoVo相比,SMSNet的性能要更好一些。由于DeepNovo没有后处理功能,为了保证比较的公平,分别展示了SMSNet在重新评分和不重新评分的情况下的性能。在下图中:
a,在由DeepNovo的作者整理的数据集上评估SMSNet和DeepNovo的氨基酸水平性能。图中标示了在5%氨基酸错误发现率下的相应召回率。
b,直方图展示了在由DeepNovo的作者整理的数据集上进行评估时,SMSNet和DeepNovo产生的置信度得分的分布。
c-d,在本文的WCU-MS-BEST数据集上进行了类似a-b的评估。
e-f,在蛋白质组学数据库获得的合成肽的高质量MS/MS谱数据集上进行了类似a-b的评估。
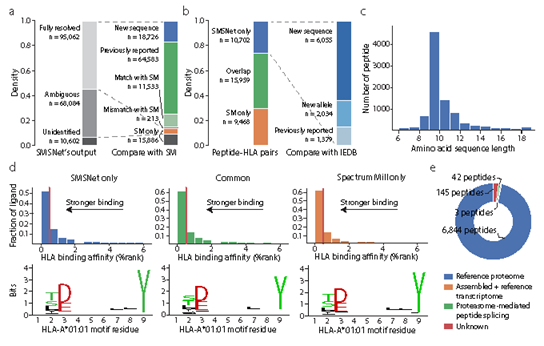
2、使用SMSNet发现了大量新的HLA抗原。在下图中:
a,使用堆叠条形图展示了由SMSNet识别的MS/MS光谱的数量以及SMSNet与先前使用Spectrum Mill软件进行MS/MS数据解释的研究之间的重叠。
b,使用堆叠条形图展示了由SMSNet识别的peptide-HLA对与免疫表位数据库(IEDB)之间的重叠。在10702个新鉴定的peptide-HLA对中,有7034种不同的肽。
c,新鉴定到的7034种肽的长度分布。
d,使用直方图和序列logos,比较了仅由SMSNet(左)、SMSNet+先前研究(中)和仅由先前研究(右)识别的peptide-HLA对之间预测的结合亲和力与核心序列motifs。
e,使用饼图展示了新鉴定的7034种肽的来源。
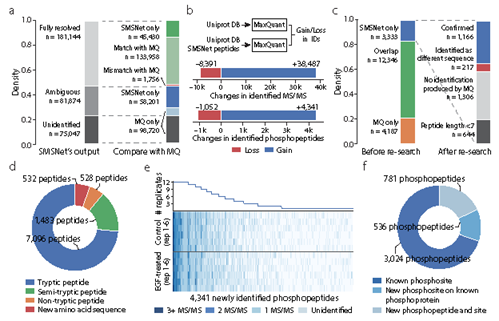
3、使用SMSNet提高了人类磷蛋白组鉴定的覆盖率。在下图中:
a,使用堆叠条形图展示了由SMSNet识别的MS/MS光谱的数量以及SMSNet与先前研究之间的重叠。
b,将SMSNet的鉴定结果加入到人类蛋白质组数据库中并用MaxQuant重新分析后,所鉴定到的MS/MS谱和磷酸肽的数量的增加和减少。
c,使用堆积条形图展示了由MaxQuant和SMSNet识别的磷酸肽数量,以及通过MaxQuant重新分析SMSNet结果从而识别到的新磷酸肽的数量。
d,饼图展示了通过将SMSNet的鉴定使用MaxQuant重新分析新鉴定到的肽的组成。这包括先前研究未鉴定的所有MS/MS光谱中的磷酸肽和非磷酸肽鉴定。
e,使用热图和线性图展示了4341个新鉴定的磷酸肽在使用MaxQuant对6个对照组和6个表皮生长因子处理的实验组进行重分析后的重现性。热图中的每一行对应一个质谱实验。
f,使用饼图展示了新鉴定的磷酸肽与PhosphoSitePlus数据库中已知的磷酸蛋白和磷酸位点之间的重叠。一个已识别的磷酸肽只有在数据库中报告了该肽上所有已识别的磷酸化位点时才被算作“Known phosphosites”。
五、文章亮点(结论讨论):本文开发的SMSNet结合了基于机器学习的现代方法和序列标记方法的优点,前者能够确定整个氨基酸序列,后者使用序列中置信度高的部分作为种子从数据库中检索完整序列,大大提升了从头测序方法的准确率。因此,SMSNet有望在新抗原发现、抗体测序和非模式生物的特征等蛋白质组学和肽组学的研究中有良好的表现。
阅读人:刘佳维
Uncovering thousands of new peptides with sequence-mask-search hybrid de novo peptide sequencing framework (使用序列掩码搜索结合肽段从头测序框架发现了数千个新肽段)-解读人:刘佳维的更多相关文章
- Mol Cell Proteomics. | Prediction of LC-MS/MS properties of peptides from sequence by deep learning (通过深度学习技术根据肽段序列预测其LC-MS/MS谱特征) (解读人:梅占龙)
通过深度学习技术根据肽段序列预测其LC-MS/MS谱特征 解读人:梅占龙 质谱平台 文献名:Prediction of LC-MS/MS properties of peptides from se ...
- 解读人:陈秋实,SP2: Rapid and Automatable Contaminant Removal from Peptide Samples for Proteomic Analyses(标准操作流程2:如何在蛋白质组学分析中快速和自动的去除肽段样品中的污染物)
发表时间:2019年4月 IF:3.950 单位: 威斯康星医学院生物化学系 威斯康星医学院生物医学质谱研究中心 物种:人(人体肾脏细胞和蛋白) 技术:肽段清理 一. 概述:(用精炼的语言描述文章的整 ...
- Analysis of endogenous peptides released from osteoarthritic cartilage unravels novel pathogenic markers (解读人:李琼)
文献名:Analysis of endogenous peptides released from osteoarthritic cartilage unravels novel pathogenic ...
- Journal of Proteome Research | SAAVpedia: identification, functional annotation, and retrieval of single amino acid variants for proteogenomic interpretation | SAAV的识别、功能注释和检索 | (解读人:徐洪凯)
文献名:SAAVpedia: identification, functional annotation, and retrieval of single amino acid variants fo ...
- 解读人:刘杰,Targeted Quantitative Kinome Analysis Identifies PRPS2 as a Promoter for Colorectal Cancer Metastasis(PRM-磷酸化激酶定量发现结肠癌转移促进因子-PRPS2)
关键词:PRM,kinase,colorectal cancer, metastasis, PRPS2 来自加州大学河滨分校的Yinsheng Wang教授应用PRM技术筛选出介导结肠癌细胞转移促进因 ...
- Sample Preparation by Easy Extraction and Digestion (SPEED) - A Universal, Rapid, and Detergent-free Protocol for Proteomics based on Acid Extraction(一种使用强酸的蛋白质提取方法SPEED,普适,快速,无需去垢剂)-解读人:李思奇
期刊名:Mol Cell Proteomics 发表时间:(2019年12月) IF:4.828 单位:德国Robert Koch 研究所 物种:多种 技术:新蛋白提取和酶解方法 一. 概述: 本文设 ...
- Journal of Proteome Research | An automated ‘cells-to-peptides’ sample preparation workflow for high-throughput, quantitative proteomic assays of microbes (解读人:陈浩)
文献名:An automated ‘cells-to-peptides’ sample preparation workflow for high-throughput, quantitative p ...
- Fast and accurate bacterial species identification in urine specimens using LC-MS/MS mass spectrometry and machine learning (解读人:闫克强)
文献名:Fast and accurate bacterial species identification in urine specimens using LC-MS/MS mass spectr ...
- Multi-batch TMT reveals false positives, batch effects and missing values(解读人:胡丹丹)
文献名:Multi-batch TMT reveals false positives, batch effects and missing values (多批次TMT定量方法中对假阳性率,批次效应 ...
随机推荐
- 吴裕雄--天生自然 R语言开发学习:广义线性模型(续一)
#----------------------------------------------# # R in Action (2nd ed): Chapter 13 # # Generalized ...
- 途牛与十八好汉撕X又言和 到底想干啥?
到底想干啥?" title="途牛与十八好汉撕X又言和 到底想干啥?"> 天下大势,合久必分,分久必合.很多看起来热闹哄哄的"劳燕分飞"事件,最 ...
- JS计算日期加天数后的日期(起始日期+有效天数=截至日期)
/** * 优惠券有效期 * startDate:起始日期 * valueTime:有效天数 */ function transferCouponValueTime(startDate,valueTi ...
- 国内游戏直播火热 为何YY游戏直播却巨亏
为何YY游戏直播却巨亏" title="国内游戏直播火热 为何YY游戏直播却巨亏"> 伴随"千万年薪主播"."美女主播陪玩那些事儿&q ...
- resin远程调试debug
wangqiaowqo Resin 远程debug Resin Windows下提升Resin默认的虚拟机内存大小 httpd.exe -Xmx1024m 参考外部文章 Resin远程debug配置文 ...
- 在不重启MySQL的情况下用gdb工具设置变量
前提:此方法只是参考其它博客的一个记录,未经亲自验证 当在mysql客户端设置一些变量时提示如下报错: 于是想能否有办法在不重启的情况下设置这些只读变量,在网上搜索别人的博客后发现如下方法 1.命令行 ...
- 2019DDCTF部分Writeup
-- re Windows Reverse1 通过DIE查壳发现存在upx,在linux上upx -d脱壳即可,拖入IDA,通过关键字符串找到关键函数: main函数中也没有什么,将输入的字符串带到s ...
- Oracle Compute云快速搭建MySQL Keepalived高可用架构
最近有个客户在测试Oracle Compute云,他们的应用需要使用MySQL数据库,由于是企业级应用一定要考虑高可用架构,因此有需求要在Oracle Compute云上搭建MySQL高可用集群.客户 ...
- ReadHub项目Kotlin版开发指南(三、MVP架构)
ReadHub项目Kotlin版转换指南(一.环境搭建) ReadHub项目Kotlin版转换指南(二.数据库和网络请求) ReadHub项目Kotlin版转换指南(三.MVP架构) Android ...
- 彻底消灭if-else嵌套
一.背景 1.1 反面教材 不知大家有没遇到过像横放着的金字塔一样的if-else嵌套: if (true) { if (true) { if (true) { if (true) { if (tru ...