NGS中的一些软件功能介绍
1.bowtie
短序列比对工具,blast也是短序列比对工具,速度快,结果易理解。
输入可以是fastq或者fasta文件。
生成比对结果文件sam格式的吧。
2.bwa
转自:https://www.jianshu.com/p/1552cc6ac3be
将DNA序列比对到参考基因组上的软件,包含三种算法:
BWA-backtrack:适合比对长度不超过100bp的序列;
BWA-SW:合于长度为70-1M bp的序列;
BWA-MEM:合于长度为70-1M bp的序列,高质量的测序数据,其比对的速度更快,精确度更高。
使用whereis bwa找到其安装路径:
xhs@dandan26:/data1/zzl$ whereis bwa
bwa: /usr/bin/bwa /usr/share/bwa /usr/share/man/man1/bwa..gz
输入bwa得到以下帮助:
Usage: bwa <command> [options] Command: index index sequences in the FASTA format
mem BWA-MEM algorithm
fastmap identify super-maximal exact matches
pemerge merge overlapping paired ends (EXPERIMENTAL)
aln gapped/ungapped alignment
samse generate alignment (single ended)
sampe generate alignment (paired ended)
bwasw BWA-SW for long queries shm manage indices in shared memory
fa2pac convert FASTA to PAC format
pac2bwt generate BWT from PAC
pac2bwtgen alternative algorithm for generating BWT
bwtupdate update .bwt to the new format
bwt2sa generate SA from BWT and Occ Note: To use BWA, you need to first index the genome with `bwa index'.
There are three alignment algorithms in BWA: `mem', `bwasw', and
`aln/samse/sampe'. If you are not sure which to use, try `bwa mem'
first. Please `man ./bwa.' for the manual.
步骤:
1.对参照基因组建索引:
bwa index –a bwtsw hg19.fasta
此处构建索引使用的是bwtsw算法,最终输出的结果文件:
会生成:bwt,pac,ann,amb,sa五种类型的文件:
xhs@dandan-PowerEdge-T620:/data1/GRCm38$ ls
GRCm38_68.fa GRCm38_68.fa.amb GRCm38_68.fa.ann GRCm38_68.fa.bwt GRCm38_68.fa.fai GRCm38_68.fa.pac GRCm38_68.fa.sa
2.使用bwa-mem算法进行比对:
bwa mem –t hg19.fasta read1.fq read2.fq > aln-pe.sam
我使用了这条命令:
bwa mem -t ../hg19/hg19.fasta ERR580012_1.fastq.gz ERR580012_2.fastq.gz > aln-pe.sam
使用了mem算法,-t是选择几个线程,增加线程,减少运行时间;然后是参照基因组的fasta文件。以及其他参数:
-p 忽略第二个输入序列,默认情况下,输入一个序列文件认为是单端测序,输入两个序列文件则是双端测序,加上这个参数后,会忽略第二个输入序列文件,把第一个文件当做单端测序的数据进行比对;
将最终结果存入到了sam文件中。
那么什么是单端测序和双端测序:
转自:https://www.cnblogs.com/Formulate0303/p/7843082.html
1、单端测序(Single-ead)首先将DNA样本进行片段化处理形成200-500p的片段,引物序列连接到DNA片段的一端,然后末端加上接头,将片段固定在flowcell上生成DNA簇,上机测序单端读取序列。
2、Paied-end方法是指在构建待测DNA文库时在两端的接头上都加上测序引物结合位点,在第一轮测序完成后,去除第一轮测序的模板链,用对读测序模块(Paied-End Module)引导互补链在原位置再生和扩增,以达到第二轮测序所用的模板量,进行第二轮互补链的合成测序。
//其实这个第二点还不太明白.[1]
3.将sam文件压缩为bam格式
samtools view –bS aln-pe_reorder.sam –o aln-pe.bam
查找samtools帮助:
Usage: samtools <command> [options] Command: view SAM<->BAM conversion
sort sort alignment file
mpileup multi-way pileup
depth compute the depth
faidx index/extract FASTA
tview text alignment viewer
index index alignment
idxstats BAM index stats (r595 or later)
fixmate fix mate information
flagstat simple stats
calmd recalculate MD/NM tags and '=' bases
merge merge sorted alignments
rmdup remove PCR duplicates
reheader replace BAM header
cat concatenate BAMs
bedcov read depth per BED region
targetcut cut fosmid regions (for fosmid pool only)
phase phase heterozygotes
bamshuf shuffle and group alignments by name
-b 表示输出为bam文件格式 –S默认下输入是 BAM 文件,若是输入是 SAM 文件,则最好加该参数,否则有时候会报错。-o 输出文件名
最终生成了bam文件,其中b指binary,运算快。
使用下面命令来查看文件头:
samtools view -H ESCell#.sam
3.下载并安装gatk
对下载的进行解压。
unzip filename.zip
将本地文件传给服务器直接使用xftp-5软件即可。
解压缩zp2类型压缩文件:
tar -xjf GenomeAnalysisTK-3.3-.tar.bz2
首次运行命令,提示:
##### ERROR MESSAGE: Fasta index file /data/data0/rawdata/../hg19/hg19.fasta.fai for reference /data/data0/rawdata/../hg19/hg19.fasta does not exist. Please see http://gatkforums.broadinstitute.org/discussion/1601/how-can-i-prepare-a-fasta-file-to-use-as-reference for help creating it.
有个关于参照组的.fai文件找不到,放进去了,然后又说没有dict文件
##### ERROR MESSAGE: Fasta dict file /data/data0/rawdata/../hg19/hg19.dict for reference /data/data0/rawdata/../hg19/hg19.fasta does not exist. Please see http://gatkforums.broadinstitute.org/discussion/1601/how-can-i-prepare-a-fasta-file-to-use-as-reference for help creating it.
5. .fasta.fai文件
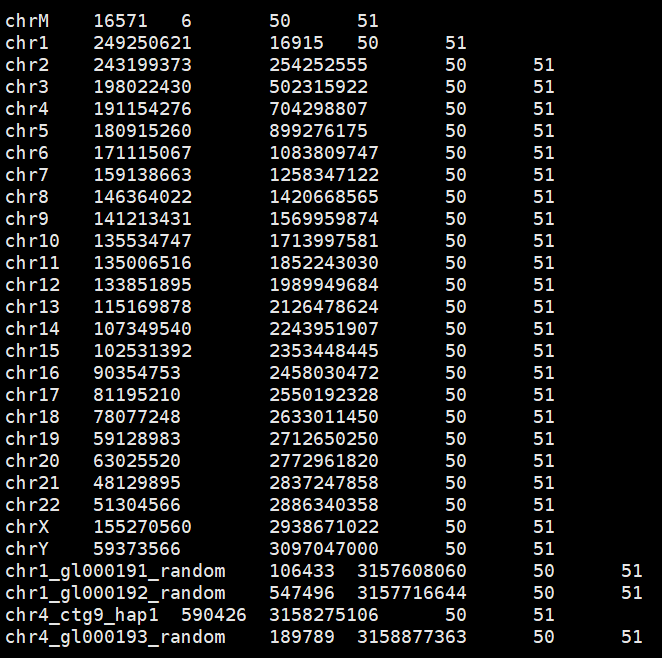
查看其中的内容,每行都有5列
name 序列长度(单位:bp) offset(第一个碱基的偏移量,从0计数,换行符也进行统计) LINEBASES (除了最后一行,其他代表序列的行的碱基数) LINEWIDTH(除了最后一行外, 其他代表序列的行的长度, 包括换行符, 在windows系统中换行符为\r\n, 要在序列长度的基础上加2)
***根据这个.fai 文件和原始的fastsa文件, 能够快速的提取任意区域的序列 。
NGS中的一些软件功能介绍的更多相关文章
- Joomla软件功能介绍与开源程序大比拼Joomla,wordpress,Drupal哪个好?
Joomla 软件功能介绍: Joomla!是一套在国外相当知名的内容管理系统 (Content Management System, CMS),它属于Portal(企业入口网站)类型,顾名思义 ...
- Android Studio 2.1及其以上版本中的instant run功能 介绍
Android Studio 2.0及其以后版本中的instant run功能 介绍 转 https://blog.csdn.net/zy987654zy/article/details/514961 ...
- sf中标准的分页功能介绍
世上本无事,庸人自扰之.我喜欢一个相对比较安静的环境去学习和工作,希望在一个掉一根针的声音都能够听到的环境中,但是有时候往往相反,一片嘈杂,我改变不了周围的环境,只能改变自己,其实这些都没有什么,也许 ...
- Python中str字符串的功能介绍
Str字符串的功能介绍 1. 字符串的操作 字符串的连接操作 符号: + 格式:str1 + str2 例如:str1 = 'I Love' str2 = 'You!' print(str1 + st ...
- 插件SimSynth合成器功能介绍
本章节采用图文结合的方式给大家介绍下电音编曲软件"水果"FL Studio中SimSynth合成器的功能介绍,感兴趣的朋友可以一起进来沟通交流哦. SimSynth插件是FL St ...
- MWeb 1.4 新功能介绍一:引入文件夹到 MWeb 中管理,支持 Octpress、Jekyll 等静态博客拖拽插入图片和实时预览
之前在 MWeb 中打开非文档库中的 Markdown 文档,如果文档中有引用到本机图片,是没办法在 MWeb 中显示出来和预览的.这是因为 Apple 规定在 Mac App Store(MAS) ...
- Autodesk Maya 2019 for Mac(三维动画软件)最新功能介绍
Autodesk Maya是美国Autodesk公司出品的世界顶级的三维动画软件,应用对象是专业的影视广告,角色动画,电影特技等.Maya功能完善,工作灵活,易学易用,制作效率极高,渲染真实感极强,是 ...
- python中列表、元组、字典内部功能介绍
一.列表(list) 常用功能的介绍:
- Python中模块之re的功能介绍
re模块的功能介绍 1. 方法 match 从开头开始查找 方法:re.match(pattern,string,flags=0) 返回值:<class '_sre.SRE_Match'> ...
随机推荐
- [mysql] Navicat for mysql_导入导出表结构
应用场景: 当 ① 由于权限控制,远程数据库在外网不能访问 ② 远程数据库连接和查询比较慢,影响工作效率 这时,可以将远程数据库的表结构和已有数据COPY到本地的mysql服务器来进行开发. 只需要将 ...
- ZooKeeper是一个分布式的,开放源码的分布式应用程序协调服务
ZooKeeper是一个分布式的,开放源码的分布式应用程序协调服务,是Google的Chubby一个开源的实现,是Hadoop和Hbase的重要组件.它是一个为分布式应用提供一致性服务的软件,提供的功 ...
- MFC通过button控制编辑框是否显示系统时间(动态显示)
1.在dlg.h中public bool flag; static UINT time(void *param); 2.在构造函数中 flag=false; 3.在button的生成函数中 if(fl ...
- VC++ Splash Window封装类CSplash
Splash.h 1234567891011121314151617181920212223242526272829303132333435363738394041424344454647484950 ...
- MySQL [Err]1449 : The user specified as a definer ('root'@'%') does not exist
权限问题:授权 给 root 所有sql 权限 mysql> grant all privileges on *.* to root@"%" identified by &q ...
- css图标
一.介绍 采用这种字体,我们可以避免网站制作中采用好多图片,一方面解决了浏览器的兼容性问题.另一方面,这些字体都是矢量字体,我们只要在调整这些图标时,将他们的字体大小以及颜色,我们就可以解决很多不是图 ...
- 采用std::thread 替换 openmp
内容转换的,具体详见博客:https://cloud.tencent.com/developer/article/1094617 及对应的code:https://github.com/cpuimag ...
- [转]C# 超高速高性能写日志 代码开源
1.需求 需求很简单,就是在C#开发中高速写日志.比如在高并发,高流量的地方需要写日志.我们知道程序在操作磁盘时是比较耗时的,所以我们把日志写到磁盘上会有一定的时间耗在上面,这些并不是我们想看到的 ...
- ios应用, 设置不自己主动备份到iCloud
原创文章,转载请注明出处 ios项目,假设有内置下载或者程序动态生成文件的话,就要注意所下载或生成的文件,要不要自己主动备份到iCloud 假设没有合适的理由就自己主动上传大文件的话,可能在不能通过应 ...
- 《C++ Primer Plus》第5章 循环和关系表达式 学习笔记
C++提供了3种循环: for 循环. while 循环 和 do while 循环 .如果循环测试条件为 true 或非零,则循环将重复执行一组指令: 如果测试条件为 false 或 0 , 则结束 ...