使用 Docker 分析高通量测序数据
端午节假期,先祝各位 Bio IT 的爱好者们,节日快乐!
做生信的童鞋想要学习 Docker,或者使用 Docker+Pipeline 封装自己的一套数据分析流程,相信一定不能错过胡博强老师在2017年写这篇《[Docker]使用阿里云 + Docker 分析高通量测序数据——RNA-Seq 与 ChIP-Seq. - Boqiang Hu》教程,这个教程同时也以推文的方式发布在了 2017-03-21 生信技能树公众号上,感兴趣的同学可以自己去翻一下。
根据教程+tangEpiNGSInstall 仓库提供的原始测试数据,本人这两天测试跑了一下,发现了一点点小问题。
$ git clone https://github.com/shenweiyan/tangEpiNGSInstall.git
$ tree
.
└── tangEpiNGSInstall
├── Dockerfile
├── README.md
├── settings
│ ├── run_chipseq.py
│ ├── run_chipseq.sh
│ ├── run_mRNA.py
│ ├── run_mRNA.sh
│ ├── scripts_chipseq.py
│ └── scripts_mRNA.py
├── src
│ └── run_sample.sh
├── test_fq
│ ├── H3K4me3
│ │ ├── test.1.fq.gz
│ │ └── test.2.fq.gz
│ ├── Input
│ │ ├── test.1.fq.gz
│ │ └── test.2.fq.gz
│ └── sample.tab.xls
└── test_fq_RNA
├── SampleA1
│ ├── test.1.fastq.gz
│ └── test.2.fastq.gz
└── sample.tab.xls
8 directories, 17 files
$ mkdir -p results database_ChIP/mm10
$ chmod 777 results database_ChIP/mm10 # avoiding Permission issue
$ tree
.
├── database_ChIP
│ └── mm10
├── results
└── tangEpiNGSInstall
├── Dockerfile
├── README.md
├── settings
│ ├── run_chipseq.py
│ ├── run_chipseq.sh
│ ├── run_mRNA.py
│ ├── run_mRNA.sh
│ ├── scripts_chipseq.py
│ └── scripts_mRNA.py
├── src
│ └── run_sample.sh
├── test_fq
│ ├── H3K4me3
│ │ ├── test.1.fq.gz
│ │ └── test.2.fq.gz
│ ├── Input
│ │ ├── test.1.fq.gz
│ │ └── test.2.fq.gz
│ └── sample.tab.xls
└── test_fq_RNA
├── SampleA1
│ ├── test.1.fastq.gz
│ └── test.2.fastq.gz
└── sample.tab.xls
11 directories, 17 files
$ docker pull hubq/tanginstall:latest
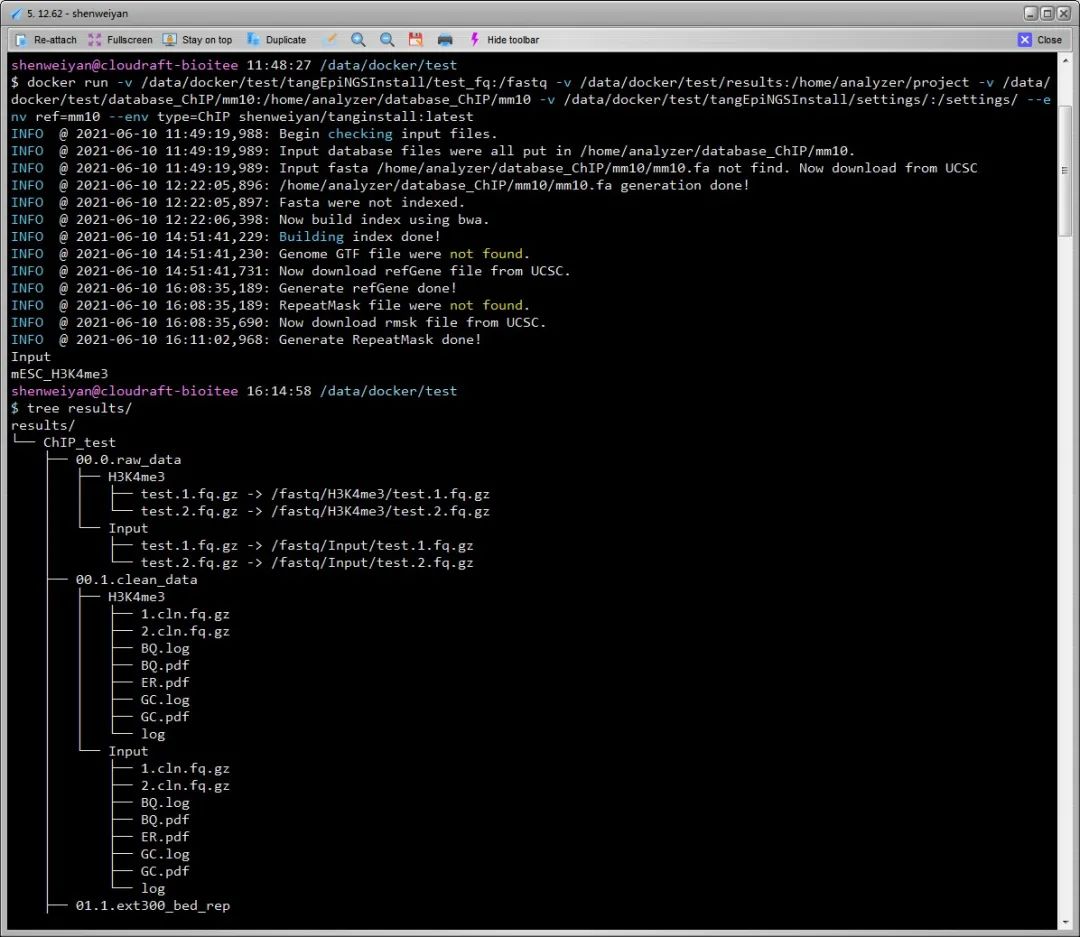
$ docker run -v /data/docker/train/tangEpiNGSInstall/test_fq:/fastq -v /data/docker/train/results:/home/analyzer/project -v /data/docker/train/database_ChIP/mm10:/home/analyzer/database_ChIP/mm10 -v /data/docker/train/tangEpiNGSInstall/settings/:/settings/ --env ref=mm10 --env type=ChIP hubq/tanginstall:latest
INFO @ 2021-06-10 03:29:48,154: Begin checking input files.
INFO @ 2021-06-10 03:29:48,154: Input database files were all put in /home/analyzer/database_ChIP/mm10.
INFO @ 2021-06-10 03:29:48,154: Input fasta /home/analyzer/database_ChIP/mm10/mm10.fa not find. Now download from UCSC
INFO @ 2021-06-10 03:45:01,604: /home/analyzer/database_ChIP/mm10/mm10.fa generation done!
INFO @ 2021-06-10 03:45:01,605: Fasta were not indexed.
INFO @ 2021-06-10 03:45:02,105: Now build index using bwa.
INFO @ 2021-06-10 03:48:20,683: Building index done!
INFO @ 2021-06-10 03:48:20,683: Genome GTF file were not found.
INFO @ 2021-06-10 03:48:21,184: Now download refGene file from UCSC.
INFO @ 2021-06-10 05:03:38,768: Generate refGene done!
INFO @ 2021-06-10 05:03:38,768: RepeatMask file were not found.
INFO @ 2021-06-10 05:03:39,269: Now download rmsk file from UCSC.
INFO @ 2021-06-10 05:06:03,592: Generate RepeatMask done!
Traceback (most recent call last):
File "/home/analyzer/module/ChIP/run_chipseq.py", line 148, in <module>
main()
File "/home/analyzer/module/ChIP/run_chipseq.py", line 126, in main
samp_peak.get_idr_stat()
File "/home/analyzer/module/ChIP/frame/module02_call_peaks.py", line 244, in get_idr_stat
mod_Stat.IDR_Stat()
File "/home/analyzer/module/ChIP/frame/module00_StatInfo.py", line 113, in IDR_Stat
f_idr_out = open(file_idr_out,"w")
IOError: [Errno 2] No such file or directory: '/home/analyzer/project/ChIP_test/StatInfo/IDR_result./home/analyzer/project/ChIP_test/sample.tab.xls'
cp: cannot stat `03.2.Peak_mrg/*/*_treat_minus_control.sort.norm.bw': No such file or directory
cp: cannot stat `03.3.Peak_idr/*/*.conservative.regionPeak.gz*': No such file or directory
cp: cannot stat `StatInfo/*': No such file or directory

出于学习和折腾,针对这个问题,个人在
hubq/tanginstall:latest 的镜像基础上做了一点小调整,并重新打包成一个名为
shenweiyan/tanginstall:latest 的新镜像 push 到了 Docker Hub,抛砖引玉,供大家学习参考。
简单说一下这个镜像的几点细节。
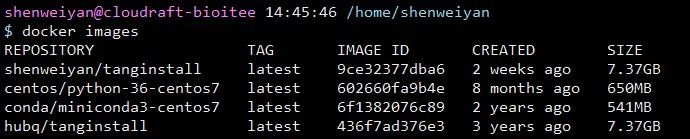
整个镜像体积比较大,总共约 7.37GB,pull 下来可能比较慢。
- 如果没有 ref(hg19/hg38 or mm9/mm10),镜像执行过程中会首先执行下载,然后拆分合并,建立 index。
db01.DownloadRef.sh
$ cat db01.DownloadRef.sh
ref=$1
dir_database=/home/analyzer/database_ChIP/$ref
dir_path=/home/analyzer/module/ChIP
cd $dir_database
wget http://hgdownload.soe.ucsc.edu/goldenPath/${ref}/bigZips/chromFa.tar.gz
tar -zxvf $dir_database/chromFa.tar.gz
for i in {1..22} X Y M
do
cat $dir_database/chr$i.fa
done >$dir_database/${ref}.fa && rm $dir_database/chr*fa
db02.RefIndex.sh
$ cat db02.RefIndex.sh
ref=$1
dir_database=/home/analyzer/database_ChIP/$ref
bwa_exe=/software/install_packages/bwa-0.7.5a/bwa
samtools_exe=/software/install_packages/samtools-0.1.18/samtools
div_bins_exe=/home/analyzer/module/ChIP/bin/div_bins/bed_read
$samtools_exe faidx $dir_database/${ref}.fa
$bwa_exe index $dir_database/${ref}.fa
$dix_bins_exe -b 100 $dir_database/${ref}.fa.fai $dir_database/columns.100.bed
$dix_bins_exe -b 1000 $dir_database/${ref}.fa.fai $dir_database/columns.1kb.bed
cut -f 1-2 $dir_database/${ref}.fa.fai >$dir_database/${ref}.fa.len
db03.RefGene.sh
$ cat db03.RefGene.sh
ref=$1
dir_database=/home/analyzer/database_ChIP/$ref
bedtools_exe=/software/install_packages/bedtools2/bin/bedtools
ucsc_dir=/software/install_packages/UCSC
bin=/home/analyzer/module/ChIP/bin
dir_path=/home/analyzer/module/ChIP
cd $dir_database
wget http://hgdownload.soe.ucsc.edu/goldenPath/${ref}/database/refGene.txt.gz
### remove chromosome fragments(unassembled).
for i in {1..22} X Y M
do
zcat $dir_database/refGene.txt.gz | grep -w chr$i
done >$dir_database/tmp
mv $dir_database/tmp $dir_database/refGene.txt
# refGene.bed
cat $dir_database/refGene.txt |\
awk '{
tag="noncoding";
if($4~/^NM/){tag="protein_coding"};
OFS="\t";
print $3,$5,$6,$2,$4,$10,$11,tag,$13
}' /dev/stdin |\
python $bin/s03_genePred2bed.py /dev/stdin |\
$bedtools_exe sort -i /dev/stdin >$dir_database/refGene.bed &&\
# region.Intragenic.bed
# For novo lncRNA detection
$bin/find_ExonIntronIntergenic/find_ExonIntronIntergenic \
$dir_database/refGene.bed \
$dir_database/${ref}.fa.fai >$dir_database/pos.bed &&\
grep -v "Intergenic" $dir_database/pos.bed |\
awk '{OFS=" ";print $1,$2,$3,"Intragenic"}' /dev/stdin \
>$dir_database/region.Intragenic.bed &&\
# refGene.gtf
# For mapping
zcat $dir_database/refGene.txt.gz |\
cut -f 2- |\
$ucsc_dir/genePredToGtf file stdin /dev/stdout |\
grep -w exon |\
$bedtools_exe sort -i /dev/stdin >$dir_database/refGene.gtf &&\
cat $dir_path/database/ERCC.gtf >>$dir_database/refGene.gtf
db04.rmsk.sh
$ cat db04.rmsk.sh
ref=$1
dir_database=/home/analyzer/database_ChIP/$ref
bedtools_exe=/software/install_packages/bedtools2/bin/bedtools
ucsc_dir=/software/install_packages/UCSC
bin=/home/analyzer/module/ChIP/bin
dir_path=/home/analyzer/module/ChIP
cd $dir_database
wget http://hgdownload.soe.ucsc.edu/goldenPath/${ref}/database/rmsk.txt.gz
zcat $dir_database/rmsk.txt.gz |\
awk '{
OFS="\t";
print $6,$7,$8,$2,".",".",".","("$9")",$10,$11,$12 "/" $13,$14,$15,$16,$17
}' /dev/stdin |\
tail -n +2 /dev/stdin >$dir_database/chrom.bed
for i in {1..22} X Y M
do
grep -w chr$i $dir_database/chrom.bed
done >$dir_database/tmp
mv $dir_database/tmp $dir_database/chrom.bed
$bedtools_exe sort -i $dir_database/chrom.bed >$dir_database/chrom.sort.bed
为节省下载时间,建议事先准备好 ${ref}.fa,如果没有,也可以先下载好以下文件。
# db01.DownloadRef.sh:
wget http://hgdownload.soe.ucsc.edu/goldenPath/${ref}/bigZips/chromFa.tar.gz
# db03.RefGene.sh:
wget http://hgdownload.soe.ucsc.edu/goldenPath/${ref}/database/refGene.txt.gz
#db04.rmsk.sh:
wget http://hgdownload.soe.ucsc.edu/goldenPath/${ref}/database/rmsk.txt.gz
bwa index(db02.RefIndex.sh)非常耗时,个人一个4核16G配置的服务器也跑了大约2.5小时。


生信服务器 | 更改 CentOS/RHEL 6/7 中的时区





本文分享自微信公众号 - 生信科技爱好者(bioitee)。
如有侵权,请联系 support@oschina.cn 删除。
本文参与“OSC源创计划”,欢迎正在阅读的你也加入,一起分享。
使用 Docker 分析高通量测序数据的更多相关文章
- NGS基础 - 高通量测序原理
NGS基础 - 高通量测序原理 原创: 赑屃 生信宝典 2017-07-23 NGS系列文章包括NGS基础.转录组分析.ChIP-seq分析.DNA甲基化分析.重测序分析五部分内容. NGS基础系列文 ...
- 【转录组入门】3:了解fastq测序数据
操作:需要用安装好的sratoolkit把sra文件转换为fastq格式的测序文件,并且用fastqc软件测试测序文件的质量 作业:理解测序reads,GC含量,质量值,接头,index,fastqc ...
- fastx_toolkit去除测序数据中的接头和低质量的reads
高通量测序数据下机后得到了fastq的raw_data,通常测序公司在将数据返还给客户之前会做"clean"处理,即得到clean_data.然而,这些clean_data是否真的 ...
- Next generation sequencing (NGS)二代测序数据预处理与分析
二代测序原理: 1.DNA待测文库构建. 超声波把DNA打断成小片段,一般200--500bp,两端加上不同的接头2.Flowcell.一个flowcell,8个channel,很多接头3.桥式PCR ...
- 单细胞转录组测序数据的可变剪接(alternative splicing)分析方法总结
可变剪接(alternative splicing),在真核生物中是一种非常基本的生物学事件.即基因转录后,先产生初始RNA或称作RNA前体,然后再通过可变剪接方式,选择性的把不同的外显子进行重连,从 ...
- linux驱动由浅入深系列:高通sensor架构实例分析之三(adsp上报数据详解、校准流程详解)【转】
本文转载自:https://blog.csdn.net/radianceblau/article/details/76180915 本系列导航: linux驱动由浅入深系列:高通sensor架构实例分 ...
- GEO(Gene Expression Omnibus):高通量基因表达数据库
Gene Expression Omnibus(GEO)是一个公共存储库,可以存档和自由分发由科学界提交的全套微阵列,新一代测序和其他形式的高通量功能基因组数据. 除数据存储外,还提供一系列基于Web ...
- 测序数据质控-FastQC
通常我们下机得到的数据是raw reads,但是公司通常会质控一份给我们,所以到很多人手上就是clean data了.我们再次使用fastqc来进行测序数据质量查看以及结果分析. fastqc的操作: ...
- 弗雷塞斯 从生物学到生物信息学到机器学习 转录组入门(3):了解fastq测序数据
sra文件转换为fastq格式 1 fastq-dump -h --split-3 也就是说如果SRA文件中只有一个文件,那么这个参数就会被忽略.如果原文件中有两个文件,那么它就会把成对的文件按*_1 ...
- 转录组入门(3):了解fastq测序数据
sra文件转换为fastq格式 fastq-dump -h --split-3 也就是说如果SRA文件中只有一个文件,那么这个参数就会被忽略.如果原文件中有两个文件,那么它就会把成对的文件按*_1.f ...
随机推荐
- Netty 心跳检测与重连机制
更多内容,前往个人博客 所谓心跳,即在 TCP 长连接中, 客户端和服务器之间定期发送的一种特殊的数据包,通知对方自己还在线,以确保 TCP 连接的有效性.心跳包还有另一个作用,经常被忽略,即:一个连 ...
- 介绍ChatGPT:基于GPT-3.5的强大自然语言处理工具
大家好,今天我们来聊一下ChatGPT,一个基于GPT-3.5架构的大型语言模型.ChatGPT在自然语言处理方面有着非常强大的能力,可以进行语言生成.文本分类.对话生成等多种任务.接下来,我们将会详 ...
- [Windows/CMD]不重启设置/刷新环境变量
1 文由 当我已经通过如下路径设置了Maven的环境(maven-3.5.4). "我的电脑"->"属性"->"高级"-> ...
- JUC(七)分支合并框架
JUC分支合并框架 简介 Fork/Join可以将一个大的任务拆分成多个子任务进行并行处理,最后将子任务的结果合并称为最终的计算结果. Fork:负责将任务拆分 Join:合并拆分任务 ForkJoi ...
- 联系我们html代码
Syntor by Aceto 11 Boleyn Court, Manor Park, Runcorn, Chesire WA7 1SR +44 (0) 1928 579865 + 44 (0) 1 ...
- FBV和CBV的区别(源码分析)
FBV和CBV源码分析 FBV直接调用user方法执行业务代码 CBV相当于在FBV上面封装了一层 from django.contrib import admin from django.urls ...
- R读入数据
两种方式: edit()自动生成一个红色的表格,列名会自动的放上去,不够的会显示var5,var6,var7 mydata <- data.frame( age = numeric(0), ge ...
- OpenAI-GPT
操作系统:CentOS 7.6 安装依赖软件 进入 root 账号: sudo -i 安装部署 ChatGPT 必备的软件,并且启动 nginx : yum install git nginx -y ...
- CS144 计算机网络 Lab4:TCP Connection
前言 经过前面几个实验的铺垫,终于到了将他们组合起来的时候了.Lab4 将实现 TCP Connection 功能,内部含有 TCPReceiver 和 TCPSender,可以与 TCP 连接的另一 ...
- dos命令、变量、字符编码、注释、用户输入
一.dos命令 1.dos命令 c: 切换盘符 cd c:\pthon 切换路径 dir 查看目录下的文件 cd .. 返回到上一层目录 cd ../.. 返回到上一层的上一层目录 二.环境变量的配置 ...