Pysam 处理bam文件
Pysam可用来处理bam文件
安装:
用 pip 或者 conda即可
使用:
Pysam的函数有很多,主要的读取函数有:
AlignmentFile:读取BAM/CRAM/SAM文件
VariantFile:读取变异数据(VCF或者BCF)
TabixFile:读取由tabix索引的文件;
FastaFile:读取fasta序列文件;
FastqFile:读取fastq测序序列文件
一般常用的是第一个和第二个。
例子:
1 import pysam
2
3 bf = pysam.AlignmentFile("in.bam","rb"); 其中r = read, b:binary. 二进制文件。 bam文件index
bf是一个迭代器,可以next()或者for读取
1 for i in bf:
2 print i.reference_name,i.pos,i.mapq,i.isize
结果:
1 ctg000331_np121 144935 27 -284
2 ctg000331_np121 144940 48 291
3 ctg000331_np121 144941 48 309
4 ctg000331_np121 144944 48 255
5 ctg000331_np121 144946 27 -370
6 ctg000331_np121 144947 27 -346
i.reference_name代表read比对到的参考序列染色体id;
- i.flag bam的flag值
i.pos代表read比对的位置;
i.mapq代表read的比对质量值;
i.isize代表PE read直接的插入片段长度,有时也称Fragment长度;
很多功能见下图:
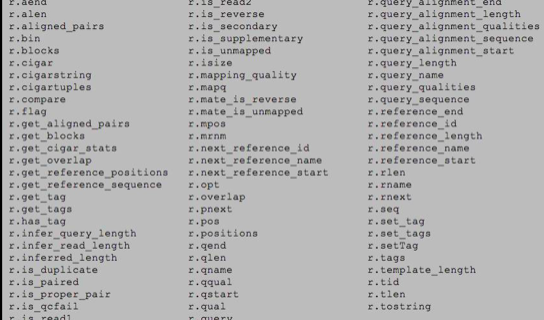
** pysam中的坐标位点是0开始,染色体起始位置为0,不是1
1 ## sam 文件依次对应的12列
2 r.qname: reads 名
3 r.flag :Flag
4 r.reference_name: 比对到的染色体
5 r.pos+1: 比对位置,必须得加一
6 r.mapq: 比对质量
7 r.cigarstring: CIGAR
8 r.next_reference_name:另外一条reads比对的参考基因组,若和第一条相同,则输出=
9 r.mpos+1: 比对的位置,必须得加1
10 r.isize: 插入片段长度
11 r.seq:reads seq
12 r.qual: reads 质量
一些功能:
- check_index()
检测index文件是否存在存在即为true
1 bf.check_index()
2 True
- close()
用完记得关闭
1 bf.close()
- count(self,contig=None, start=None, stop=None, region=None, until_eof=False, read_callback='nofilter', reference=None,end=None)
计算目标区域内比对上的reads数目
1 bf.count(contig="ctg000331_np121", start=1, stop=6000)
2 24
- count_coverage(self, contig=None, start=None, stop=None, region=None, quality_threshold=15, read_callback='all', reference=None, end=None)
计算目标区域内的覆盖度。返回1个4维的array,代表ACGT的覆盖度,而每个维度的array长度为100,里面的数字代表该碱基在各个位置上的覆盖度。
1 bf.count_coverage(contig="ctg000331_np121",start=1,stop=100)
- fetch(self, contig=None, start=None, stop=None, region=None, tid=None, until_eof=False, multiple_iterators=False, reference=None, end=None)
提取出比对到目标区域内的全部reads。返回的是一个迭代器,可以通过for循环或者next函数从中取出reads,我们使用next()函数取出第一条reads,reads是用 AlignedSegment对象表示,可以通过该对象的内置方法再对这条reads进行一些查询操作。
1 allreads=bf.fetch(contig="ctg000331_np121",start=1,stop=10000)
2 是一个迭代器,可以用for循环获得
- get_index_statistics(self)
通过index统计该BAM文件中在各个染色体上mapped/unmapped的reads个数
1 bf.get_index_statistics()
- fetch函数定位特定区域
有时候我们并不需要遍历整一份BAM文件,我们可能只想获得区中的某一个区域(比如chr1中301-310中的信息),那么这个时候可以用Alignmen模块中的fetch函数:
bam文件必须要index
1 for r in bf.fetch('chr1', 300, 310):
2 print r
3 bf.close()
关注下方公众号可获得更多精彩

参考
Pysam 处理bam文件的更多相关文章
- pysam操作sam文件
pysam模块 因为要分析sam文件中序列的情况,因此要对reads进行细分,所以之前想用数据库将sam文件信息存储,然后用sql语句进行分类.后来发现很麻烦,pysam就是一个高效读取存储在SAM ...
- SAM/BAM文件处理
当测序得到的fastq文件map到基因组之后,我们通常会得到一个sam或者bam为扩展名的文件.SAM的全称是sequence alignment/map format.而BAM就是SAM的二进制文件 ...
- bam文件softclip , hardclip ,markduplicate的探究
测序产生的bam文件,有一些reads在cigar值里显示存在softclip,有一些存在hardclip,究竟softclip和hardclip是怎么判断出来的,还有是怎么标记duplicate ...
- C++使用htslib库读入和写出bam文件
有时候我们需要使用C++处理bam文件,比如取出read1或者read2等符合特定条件的序列,根据cigar值对序列指定位置的碱基进行统计或者对序列进行处理并输出等,这时我们可以使用htslib库 ...
- SAMTOOLS使用 SAM BAM文件处理
[怪毛匠子 整理] samtools学习及使用范例,以及官方文档详解 #第一步:把sam文件转换成bam文件,我们得到map.bam文件 system"samtools view -bS m ...
- bam文件测序深度统计-bamdst
最近接触的数据都是靶向测序,或者全外测序的数据.对数据的覆盖深度及靶向捕获效率的评估成为了数据质量监控中必不可少的一环. 以前都是用samtools depth 算出单碱基的深度后,用perl来进行深 ...
- 文件格式——Sam&bam文件
Sam&bam文件 SAM是一种序列比对格式标准, 由sanger制定,是以TAB为分割符的文本格式.主要应用于测序序列mapping到基因组上的结果表示,当然也可以表示任意的多重比对结果.当 ...
- 推荐一个SAM文件或者bam文件中flag含义解释工具
SAM是Sequence Alignment/Map 的缩写.像bwa等软件序列比对结果都会输出这样的文件.samtools网站上有专门的文档介绍SAM文件.具体地址:http://samtools. ...
- 怎么从bam文件中提取出比对OR没比对上的paired reads | bamToFastq | STAR
折腾这么多都是白瞎,STAR就有输出没有别对上的pair-end reads的功能 参见:How To Filter Mapped Reads With Samtools I had the same ...
随机推荐
- Sequence Model-week2编程题1-词向量的操作【余弦相似度 词类比 除偏词向量】
1. 词向量上的操作(Operations on word vectors) 因为词嵌入的训练是非常耗资源的,所以ML从业者通常 都是 选择加载训练好 的 词嵌入(Embedding)数据集.(不用自 ...
- [no code][scrum meeting] Alpha 9
项目 内容 会议时间 2020-04-15 会议主题 OCR验收 会议时长 15min 参会人员 OCR组成员 $( "#cnblogs_post_body" ).catalog( ...
- Noip模拟45 2021.8.21
一定别删大括号,检查是;还是, ceil函数里面要写double,否则根本没用!!!!!!! T1 打表 正解:打表 考场上很难真正把柿子理解着推出来 况且想要理解题意就很难,比如我就理解错了 半猜着 ...
- 使用jQuery-UI来实现一个Ajax的自动完成功能(自动填充搜索框的下拉值)
首先你要在.net拓展包中去搜索 jquery ui (Combined Libray)安装这么个文件 第二部 在控制器中添加我们根据输入搜索框的值获取符合的记录集的action 第三步 有了 ...
- Python import cStringIO ImportError: No module named 'cStringIO'
From Python 3.0 changelog; The StringIO and cStringIO modules are gone. Instead, import the io modul ...
- 网关服务spring cloud zuul
Zuul例子配置文件 spring.application.name=switch-gateway server.port=5555 请求路由 传统路由方式 zuul.routes.api-a-url ...
- k8s入坑之路(7)kubernetes设计精髓List/Watch机制和Informer模块详解
1.list-watch是什么 List-watch 是 K8S 统一的异步消息处理机制,保证了消息的实时性,可靠性,顺序性,性能等等,为声明式风格的API 奠定了良好的基础,它是优雅的通信方式,是 ...
- 解决mac主机无法与 Docker容器互通问题
方法很多,这里我说一下使用 docker-connector解决这个问题 这是一个github开源项目docker-connector 1. Mac 通过 brew 安装 docker-connec ...
- java中使用Process执行linux命令
代码如下 BufferedReader reader = null; String cmd = "netstat -anp|grep :8080";//命令中有管道符 | 需要如下 ...
- BAT面试必问细节:关于Netty中的ByteBuf详解
在Netty中,还有另外一个比较常见的对象ByteBuf,它其实等同于Java Nio中的ByteBuffer,但是ByteBuf对Nio中的ByteBuffer的功能做了很作增强,下面我们来简单了解 ...