bam文件softclip , hardclip ,markduplicate的探究
测序产生的bam文件,有一些reads在cigar值里显示存在softclip,有一些存在hardclip,究竟softclip和hardclip是怎么判断出来的,还有是怎么标记duplicate的reads的,我怀着这些问题进行了探究。
测试步骤
- 编辑两个bed文件,分别含有我们需要的read1和read2位置,这里每个文件包含两条read1或者两条read2,read1、read2一对作为原始的reads(序列名primer_pri),另一对作为截取的材料(这里取序列名为other)
- 使用bedtools getfasta,从参考基因组获得reads的序列,将read2反向互补。将原始reads放入两个文件,一个test_R1.fa,一个test_R2.fa
在test_R1.fa中添加其它修改过的原始reads,并在test_R2.fa中也添加相应的read2,不过read2不修改
read1名称如下

- primer_pri:原始read
- pimer_duplicate1:primer_pri的重复,一模一样
- pimer_duplicate2:read1 primer_pri去掉5‘两个碱基
- pimer_duplicate3:read1 primer_pri去掉5’两个碱基,再去掉3'两个碱基
- pimer_changeR2Termi5base:read1修改了read2 5‘端的碱基
- primer_halfother:read1截掉后面reads,用other的5‘部分reads补全
- pimer_change3Termi5base_change5Termi5base_sametwo:read1和read2一样,并且5'端和3‘端都改变了5个碱基
pimer_change3Termi5base_change5Termi5base:read1 5'端和3‘端都改变了5个碱基,但是read2保留primer_pri的read2
结果
softclip和hardclip
其中
- primer_halfother read1 82M65S,有SA tag,SA:Z:chr12,5378700,+,79S68M,60,0
- pimer_change3Termi5base_change5Termi5base_sametwo 两条reads均为5S137M5S
- pimer_change3Termi5base_change5Termi5base read1 5S137M5S
- primer_halfother read1 79H68M ,有SA tag,SA:Z:chr12,5378502,+,82M65S,60,0
结论(部分分析参考SAMv1.pdf文件)
- 对于map到一个位置的read,两端map不上的叫做clip ,map到一个位置的情况下以softclip显示(比如 pimer_change3Termi5base_change5Termi5base_sametwo和 pimer_change3Termi5base_change5Termi5base read1)
- 对于嵌合比对的read(可以map到多个区域,并且这比对上的区域很大部分非overlap),比如primer_halfother read1比对上两个位置,一个比对到chr12 : 5378502,一个比对到chr12:5378700,并且两次hit的位置的碱基overlap少,产生的这种情况是因为read前一部分比对到了前者,而后一部分又可以比对到了后者,因此无论比对到任何一个位置都这条read都是部分match(这种叫做Chimeric alignment/嵌合比对)。
- 嵌合比对的read,有一条是最优的read,因为我们map的时候设置了-M参数,因此认为较短的split的reads断定为优,这里是的62 clip 的hit断定为优。因此65个比对不上的显示为softclip,而另外一个hit,79 clip显示为hard clip,序列中不显示,并且存入0x800(supplementary alignment flag)
- 为什么82M65S对应的是79H68M呢,理论上应该是82H65M才对,这是因为这里两个比对有三个碱基的overlap,所以前面有65+3个match,后面有79+3个match(制造reads的时候碰巧截取的primer read 3'端三个碱基和截取的other read 5‘部分read 三个碱基一样)
- 这种嵌合比对的reads含有SA tag
duplicate
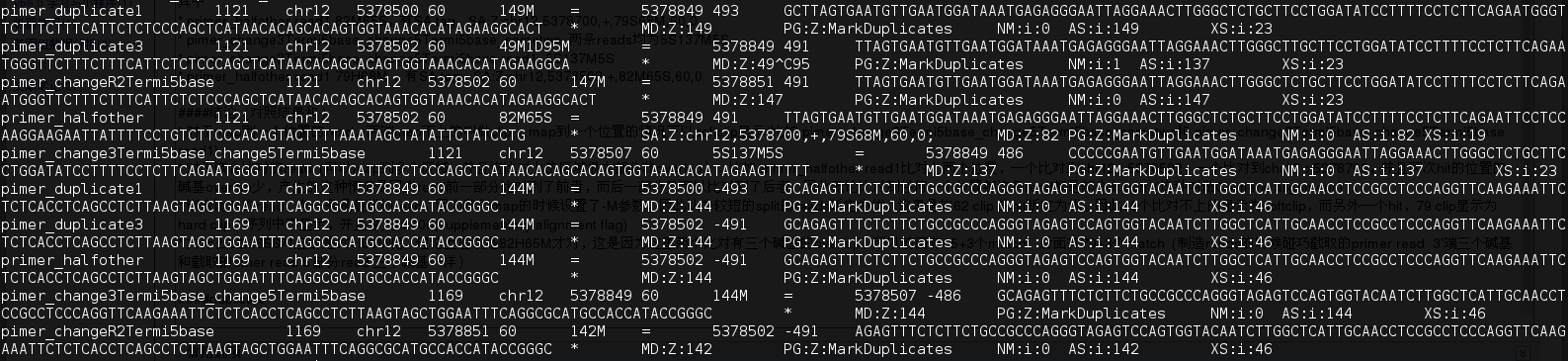
其中mark为duplicate 的reads 对(duplicate 是按fragment算) 有 primer duplicate1,primer_duplicate3,pimer_changeR2Termi5base,primer_halfother(82M65S,144M(未改的read2)),pimer_change3Termi5base_change5Termi5base
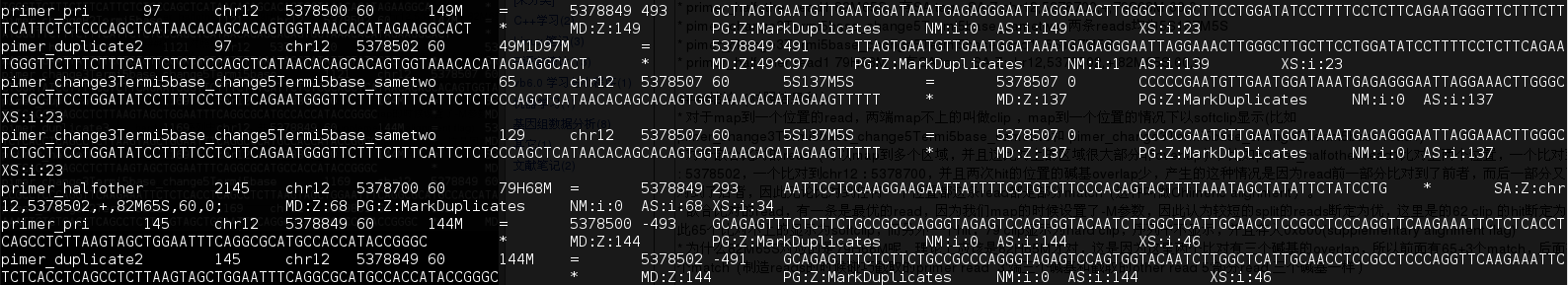
不属于duplicate的有
primer_pri,pimer_duplicate2,primer_halfother(79H68M,一条),pimer_change3Termi5basechange5Termi5base_sametwo
结论
- fragment的start和end一样(read1和read2(因为read2是测对链的,reads的5‘端都是fragment的末端)的5’的位置都相同)判断为duplicate,只取最优的不标记为duplicate
- primer_pri的duplicate是 primer duplicate1, primer_halfother
- pimer_duplicate2的duplicate是primer_duplicate3,pimer_changeR2Termi5base,pimer_change3Termi5base_change5Termi5base
- 没有duplicate的是primer_halfother(79H68M,一条),pimer_change3Termi5basechange5Termi5base_sametwo
- pimer_change3Termi5basechange5Termi5base_sametwo 5'有5 softclip,map的位置从M的碱基算起(见图),所以它没有duplicate
bam文件softclip , hardclip ,markduplicate的探究的更多相关文章
- SAMTOOLS使用 SAM BAM文件处理
[怪毛匠子 整理] samtools学习及使用范例,以及官方文档详解 #第一步:把sam文件转换成bam文件,我们得到map.bam文件 system"samtools view -bS m ...
- SAM/BAM文件处理
当测序得到的fastq文件map到基因组之后,我们通常会得到一个sam或者bam为扩展名的文件.SAM的全称是sequence alignment/map format.而BAM就是SAM的二进制文件 ...
- C++使用htslib库读入和写出bam文件
有时候我们需要使用C++处理bam文件,比如取出read1或者read2等符合特定条件的序列,根据cigar值对序列指定位置的碱基进行统计或者对序列进行处理并输出等,这时我们可以使用htslib库 ...
- Vivado约束文件(XDC)的探究(2)
Vivado约束文件(XDC)的探究(2)
- Vivado约束文件(XDC)的探究(1)
Vivado约束文件(XDC)的探究(1) 工程建好之后会出现xdc文件: 注意:active 和 target 生成的约束文件如下:
- bam文件测序深度统计-bamdst
最近接触的数据都是靶向测序,或者全外测序的数据.对数据的覆盖深度及靶向捕获效率的评估成为了数据质量监控中必不可少的一环. 以前都是用samtools depth 算出单碱基的深度后,用perl来进行深 ...
- 文件格式——Sam&bam文件
Sam&bam文件 SAM是一种序列比对格式标准, 由sanger制定,是以TAB为分割符的文本格式.主要应用于测序序列mapping到基因组上的结果表示,当然也可以表示任意的多重比对结果.当 ...
- Pysam 处理bam文件
Pysam可用来处理bam文件 安装: 用 pip 或者 conda即可 使用: Pysam的函数有很多,主要的读取函数有: AlignmentFile:读取BAM/CRAM/SAM文件 Varian ...
- 推荐一个SAM文件或者bam文件中flag含义解释工具
SAM是Sequence Alignment/Map 的缩写.像bwa等软件序列比对结果都会输出这样的文件.samtools网站上有专门的文档介绍SAM文件.具体地址:http://samtools. ...
随机推荐
- 【D3】transition API
摘要: 动画类API 一.API 使用 1. 1 d3.ease 1.2 d3.timer Start a custom animation timer, invoking the specified ...
- 用letsencrypt搭建免费的https网站
环境:阿里云服务器centos7.3,nignx,letsencrypt做免费的https证书 Let’s Encrypt官网:https://letsencrypt.org/ 1.服务器开放端口:4 ...
- 百度地图api将可视区域定位到当前所在位置
1.前言 开头不说点什么,总是有点不习惯.还是说点什么吧,关于百度地图,我用的次数还是比较多的,没办法,需求呀.好吧,在用百度地图的时候,确实有过很多需求,不过好在百度地图很强大,每次需求在探索后都能 ...
- 支付宝分库分表中间件--zdal简介
中间件, 如果仅仅作为一名用户的话, 主要关注一下如何使用即可, 大多数情况下也就是配置. 下面简单的介绍一下支付宝的分库分表中间件--->zdal在web项目中的配置. 1, 在网上查阅相关资 ...
- 蓝桥杯比赛javaB组练习《饮料换购》
题目如下: 饮料换购 乐羊羊饮料厂正在举办一次促销优惠活动.乐羊羊C型饮料,凭3个瓶盖可以再换一瓶C型饮料,并且可以一直循环下去,但不允许赊账. 请你计算一下,如果小明不浪费瓶盖,尽量地参加活动,那么 ...
- An internal error occurred during: "Launching web on MyEclipse Tomcat"
An internal error occurred during: "Launching web on MyEclipse Tomcat" 解决办法1 1.首先关闭MyEclip ...
- 【Lab】提取result的bits和Y-PSNR数据并整理到Excel
[Lab]提取result的bits和Y-PSNR数据并整理到Excel 更新:使用openpyxl库直接将数据写入Excel中 注意:openpyxl是第三方库,如果没有安装.请命令行里键入pip ...
- 设计模式笔记——GoF设计模式汇总
目录 · 总述 · 记忆 · 效果 · 面向对象设计原则 · 创建型模式 · 单例模式(Singleton) · 效果 · 分类 · 代码(饿汉式) · 代码(懒汉式) · 代码(双重检测锁式) · ...
- css的选择器的优先级
css覆盖是在打代码的时候,开发者很普通很普通,也是很经常经常用到的,但是容易混淆他们之间的优先级. [][][] 第一个是id,第二个是class,第三个是元素名.有一个就加一.比较这个三位数的大小 ...
- Oracle 11.2.0.1 ADG环境MRP进程遭遇ORA-600异常终止
环境:Linux + Oracle 11.2.0.1 ADG 现象:发现备库没有应用日志 1. 数据库查询备库目前状态 发现备库目前没有应用日志,apply lag已经显示备库有3天21小时多没有应用 ...