用R包中heatmap画热图
一:导入R包及需要画热图的数据
library(pheatmap)
data<- read.table("F:/R练习/R测试数据/heatmapdata.txt",head = T,row.names=1,sep="\t")
二:画图
1)pheatmap(data)#默认参数
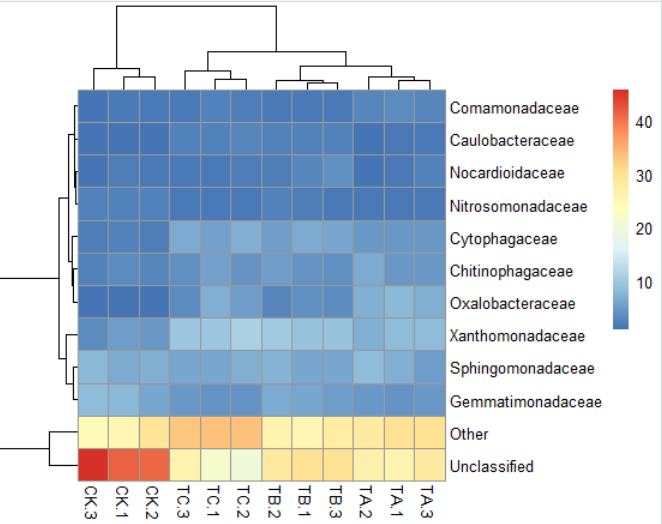
2)pheatmap(data,clustering_distance_rows = "correlation")#聚类线长度优化

3)pheatmap(data,scale="column")#按列均一化,"row","column" or "none"默认是"none"
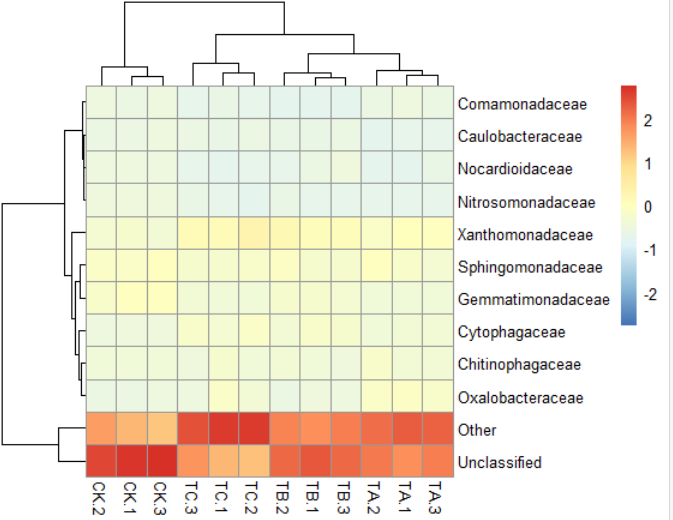
4)pheatmap(data,scale="row")#按行均一化,"row","column" or "none"默认是"none"
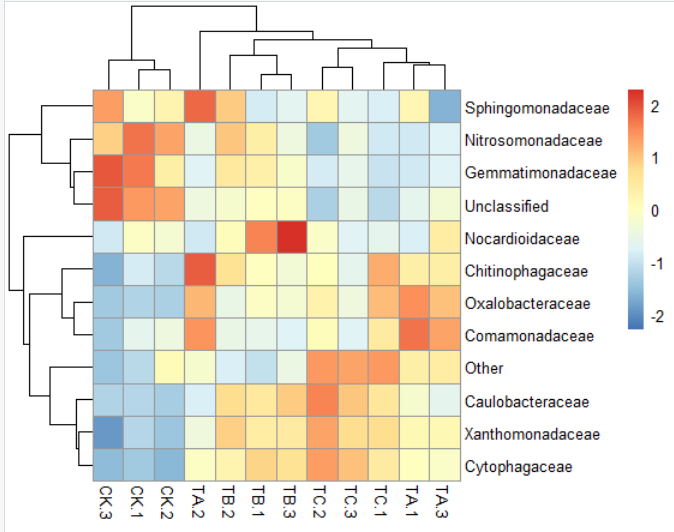
5)pheatmap(data,display_numbers=T,number_format="%.2f",number_color="red",fontsize_number=8)#是否在每一格上显示数据,及其数据格式,大小及其颜色
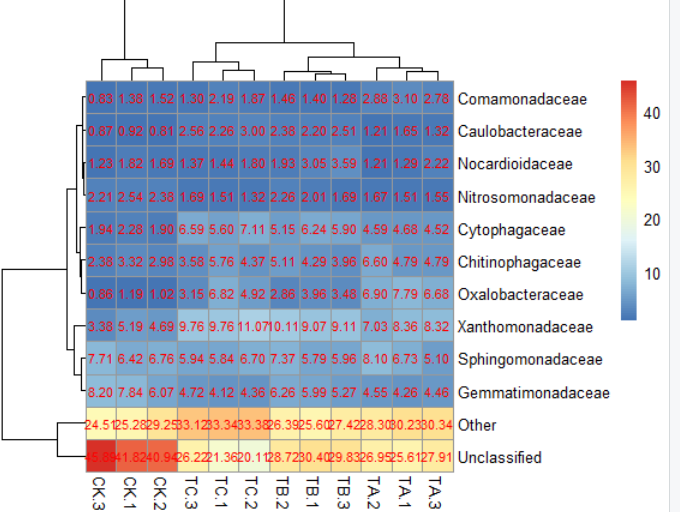
6)pheatmap(data,cellwidth = 50,cellheight= 14)#格子大小
7) pheatmap(data,main="sbheatmap")#标题
8)pheatmap(data,color = colorRampPalette(c("MediumBlue","white","red"))(256))#颜色
9)pheatmap(data,clustering_distance_rows = "correlation",scale="column",display_numbers=T,number_format="%.1f",number_color="black",
fontsize_number=8,cellwidth = 14,cellheight= 14,color = colorRampPalette(c("MediumBlue","white","red"))(256))

原因是当每行只有两个数值的时候,任何两个不同的数值标准正态均一化后,都会变成-1和1。所以,会产生上述的图形(只有两种颜色)。面对这种类型的数据,建议直接计算每行两个数值的倍数的log2值,然后使用OS-tools画单列的热图。
当然, 单列热图也可以使用R语言的pheatmap包绘制,并通过一个函数控制0点的位置,在另一个R语言绘图的主题帖中也有介绍。
(3)聚类的原理
如同上图的聚类效果,简单说来,在基因完成归一化处理后,如果我们对行聚类(在上图中也就是对基因聚类),那么基因的顺序就会被重排。表达规律比较相似的基因将会被排在一起。表达模式差异越大的基因,则会远离。类似的,如果对列聚类(在上图中也就是对样本聚类),那么样本的顺序将会被重排,表达模式比较相似的样本自动会被归为一类。
例如,在以下的热图中,选择了对行(基因)聚类,不对列(样本)聚类。那是因为作者希望通过聚类,将表达模式相似基因归为一类在图中展示,所以基因聚类选择yes。而样本(列)是作者提前排好序的,是小鼠三个组织在6个发育阶段的样本。因为样本是提前排好序的,当然作者不希望这个顺序被打乱,所以列选择不聚类。
还有两个方向都不聚类的例子。例如在下图中,X轴是1个实验处理后0h、5h、10h的样本,是作者提前排好序的。本意是想呈现相关基因在梯度时间水平的变化规律。当然,作者不想这个顺序被重新排布了,所以列方向的聚类选择:no。在y轴方向,这些基因也是作者提前按照其所属的基因家族排过序的,当然也不想其顺序被打乱,所以行聚类也是选择no。
用R包中heatmap画热图的更多相关文章
- R绘图(2): 离散/分类变量如何画热图/方块图
相信很多人都看到过上面这种方块图,有点像"华夫饼图"的升级版,也有点像"热图"的离散版.我在一些临床多组学的文章里面看到过好几次这种图,用它来展示病人的临床信息 ...
- 机器学习数据集,主数据集不能通过,人脸数据集介绍,从r包中获取数据集,中国河流数据集
机器学习数据集,主数据集不能通过,人脸数据集介绍,从r包中获取数据集,中国河流数据集 选自Microsoft www.tz365.Cn 作者:Lee Scott 机器之心编译 参与:李亚洲.吴攀. ...
- Matplotlib学习---用matplotlib画热图(heatmap)
这里利用Nathan Yau所著的<鲜活的数据:数据可视化指南>一书中的数据,学习画图. 数据地址:http://datasets.flowingdata.com/ppg2008.csv ...
- HeatMap(热图)的原理和实现
先来看两张图: (1)10年世界杯决赛,冠军西班牙队中门将.后卫.中场及前锋的跑位热图 通过热图,我们可以很清楚的看出四个球员在比赛中跑动位置的差异. (2)历史地震震源位置的热图 也可以很清楚的看出 ...
- python中matplotlib画折线图实例(坐标轴数字、字符串混搭及标题中文显示)
最近在用python中的matplotlib画折线图,遇到了坐标轴 "数字+刻度" 混合显示.标题中文显示.批量处理等诸多问题.通过学习解决了,来记录下.如有错误或不足之处,望请指 ...
- 【代码更新】单细胞分析实录(20): 将多个样本的CNV定位到染色体臂,并画热图
之前写过三篇和CNV相关的帖子,如果你做肿瘤单细胞转录组,大概率看过: 单细胞分析实录(11): inferCNV的基本用法 单细胞分析实录(12): 如何推断肿瘤细胞 单细胞分析实录(13): in ...
- seaborn用heatmap画热度图
原文链接 https://blog.csdn.net/m0_38103546/article/details/79935671
- complexHeatmap包画分类热图
用途:一般我们画热图是以连续变量作为填充因子,complexHeatmap的oncopoint函数可以以类别变量作为填充因子作热图. 用法:oncoPrint(mat, get_type = func ...
- gplots heatmap.2和ggplot2 geom_tile实现数据聚类和热图plot
主要步骤 ggplot2 数据处理成矩阵形式,给行名列名 hclust聚类,改变矩阵行列顺序为聚类后的顺序 melt数据,处理成ggplot2能够直接处理的数据结构,并加上列名 ggplot_tile ...
随机推荐
- WPF c# 定时器
//定时查询-定时器 DispatcherTimer dispatcherTimer = new DispatcherTimer(); dispatcherTimer.Tick += (s, e) = ...
- concurrent.futures模块 -----进程池 ---线程池 ---回调
concurrent.futures模块提供了高度封装的异步调用接口,它内部有关的两个池 ThreadPoolExecutor:线程池,提供异步调用,其基础就是老版的Pool ProcessPoolE ...
- Vue.js基础(二)
属性 可以将数据进行计算得出新的结果,也可以说是一个公式,有缓存. 应用:1,频繁使用的复杂公式. 2,需要监控的-----全局状态的管理 简写: computed:{ result:functi ...
- tkinter面板切换
- 2.HTML文件中<!DOCTYPE html>的作用
<!DOCTYPE> 声明位于文档中的最前面的位置,处于 <html> 标签之前.此标签可告知浏 览器文档使用哪种 HTML 或 XHTML 规范.(重点:告诉浏览器按照何种规 ...
- spring 之 factory-bean & factory-method
这两者常常是一起出现的,或者说他们经常是一起被使用的.但是其实是分为了两种情况: 1 同时使用factory-bean 和 factory-method 如果,我们在一个bean 元素上同时配置 fa ...
- Maven的下载和配置
一.下载 打开 链接地址 http://maven.apache.org/download.cgi 下载 Maven, 下载加压打开以后目录: 二.配置环境变量 我的电脑 -> 属性->高 ...
- hive 修复元数据命令 & 如何快速复制一张hive的分区表
hive 元数据修复命令 msck repair table xxx; 也可以用于分区表的快速复制 例如你需要从线上往线下导一张分区表,但是网又没有连通,你需要如何操作呢? 1.复制建表语句 2.从线 ...
- leetcode1003
class Solution: def isValid(self, S: str) -> bool: n = len(S) if n % 3 != 0: return False while n ...
- 【转】簡單講講 USB Human Interface Device
原地址http://213style.blogspot.com/2013/09/usb-human-interface-device.html 恩,發本文的原因是看到了以前畢業的朋友在旁邊的對話框問了 ...