Repbase library|divergence rate|self-sequence alignment|genomic rearrangement|cutoffs|breakpoint
(Panda, dog and human repeat comparison):与其他动物比较重复序列
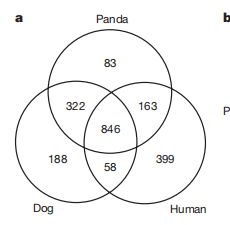
我们使用Repbase 库(重复序列库)+已知的转录原件序列+识别软件,评估出转录原件占比,并且与狗和人相比。用Repbase数据库(扩张度来自repeat base)分析熊猫基因中的转录原件的扩张度,得到:大部分熊猫转录原件基因有超过10% 的共有序列的扩张度(这是因为repbase数据库数据基于哺乳动物基因,并没有大熊猫基因)。小部分低于10%的扩张度(这可能是哺乳动物最近起源的活性转座子)。
Repbase library:database mask and annotation repetitive DNA
RepeatModeller:de-novo repeat family identification and modeling package
(Panda genome has a low divergence rate):
(investigate the rate of recent segmental duplication):采用self-sequence alignment,先在全基因组识别出重复片段个数及其大小(因为测序reads与组装的全基因组相比较,测序readsmapping上的重复片段多,所以在assembly过程中丢失了一些重复片段,同时因为这些片段的reads深度明显高于其他区域,所以要与depth相乘),所以根据平均depth(重复片段的average depth)和测序reads大小,得到重复片段的大小。
self-sequence alignment:自己的基因和自己的基因相互比对得到
(investigate panda genome conservation and evolution,)比对了狗,人,熊猫的全基因组,三者有相似序列,不相似的部分:包括狗与熊猫的相似度高于(人与狗,人与熊猫),熊猫的特异性序列最少,所以熊猫基因组的扩展性是最低的。
(the panda, dog and human genomes had high genomic synteny)比人,狗,熊猫第二条染色体的35条scaffold,没发现大规模重排。conserved synteny+pairwise syntenic regions
conserved synteny:保守同源区;pairwise syntenic regions:成对同源区
(genomic rearrangement events)重排事件,用几乎全基因组的scaffold序列,比较狗和熊猫的染色体间较小同源片段,片段有大有小(之前打断成不同大小的小序列),使用人类基因作为参照,发现dog中重排是panda的三倍,所以表明panda的扩张度较低。
Cutoff:截取片段
Repbase library|divergence rate|self-sequence alignment|genomic rearrangement|cutoffs|breakpoint的更多相关文章
- Multiple sequence alignment Benchmark Data set
Multiple sequence alignment Benchmark Data set 1. 汇总: 序列比对标准数据集: http://www.drive5.com/bench/ This i ...
- [Sequence Alignment Methods] Dynamic time warping (DTW)
本系列介绍几种序列对齐方法,包括Dynamic time warping (DTW),Smith–Waterman algorithm,Cross-recurrence plot Dynamic ti ...
- [Sequence Alignment Methods] Cross-Recurrent Plot (CRP)
A recurrence plot (RP) is a straightforward way to visualize characteristics of similar system state ...
- [Sequence Alignment Methods] Smith–Waterman algorithm
Smith–Waterman algorithm 首先需要澄清一个事实,Smith–Waterman algorithm是求两个序列的最佳subsequence匹配,与之对应的算法但是求两个序列整体匹 ...
- The sequence and de novo assembly of the giant panda genome.ppt
sequencing:使用二代测序原因:高通量,短序列 不用长序列原因: 1.算法错误率高 2.长序列测序将嵌合体基因错误积累.嵌合体基因:通过重组由来源与功能不同的基因序列剪接而形成的杂合基因 se ...
- 使用IDENTITY列属性和Sequence对象
使用IDENTITY列属性 1. 建立表 Sales.MyOrders USE TSQL2012; IF OBJECT_ID(N'Sales.MyOrders', N'U') IS NOT NULL ...
- 下载并安装Prism5.0库 Download and Setup Prism Library 5.0 for WPF(英汉对照版)
Learn what’s included in Prism 5.0 including the documentation, WPF code samples, and libraries. Add ...
- GATK-BWA-MEM handle GRCh38 alternate contig mappings
1. For the Impatient # Download bwakit (or from <http://sourceforge.net/projects/bio-bwa/files/bw ...
- SAMTOOLS使用 SAM BAM文件处理
[怪毛匠子 整理] samtools学习及使用范例,以及官方文档详解 #第一步:把sam文件转换成bam文件,我们得到map.bam文件 system"samtools view -bS m ...
随机推荐
- KM算法萌新讲解篇
KM算法 首先了解问题:也就是最大权值匹配: 二分图里,边带了权值,求整幅图里匹配最大/最小的权值 因为接触匈牙利算法的时候看的是找对象系列的博文,所以也自己写一发找对象的博文吧: 算法背景: 信 ...
- Aandroid 解决apk打包过程中出现的“Certificate for <jcenter.bintray.com> doesn't match any of the subject alternative names: [*.aktana.com, aktana.com]”的问题
有时候,apk打包过程中会出现“Certificate for <jcenter.bintray.com> doesn't match any of the subject alterna ...
- C#连接Sqlite实现单表操作
今天我们来了解下VS使用的众多数据库中比较轻量的数据库SQLITE,好处当然就在于“轻~”!!!.自己理解
- BZOJ2152聪聪可可
bzoj传送门 luogu传送门 这题算是很sb的点分治了,最近在点分治复习,写了练练手,对于这个题只需要对统计0,1,2出现的次数就好了吧,然后发现答案不对,也就是每个点对需要算两遍嘛,0也算,所以 ...
- Mysql 开启 Slow 慢查询
1:登录数据库查看是否已经开启了Slow慢查询: mysql> show variables like 'slow_query%'; 2:开启Mysql slow日志: 默认情况下slow_qu ...
- PostgreSQL-7-数据连接
1.通过WHERE进行简单连接 SELECT * FROM company3,department 不添加WHERE将会显示所有数据 SELECT * FROM company3,departmen ...
- C# 面向对象之3个基本特征
C#是面向对象的语言,每个面向对象语言都有3个基本特征: *封装----把客观的事物封装成类,并将类的内部实现隐藏,以保证数据的完整性. *继承----通过继承可以复用父类的代码. *多态----允许 ...
- Lock简介
digest synchronized已经提供了锁的功能,而且还是Java的内置特性,那为什么还要出现lock呢? 用一句话来讲就是——synchronized可以实现同步,但太死板了它的同步机制:l ...
- NUP2201MR
NUP2201MR:双总线保护IC(瞬态抑制二极管),常用于USB总线的保护.
- 创建Maven项目出错 pom出错
错误为 org.apache.maven.archiver.MavenArchiver.getManifest(org.apache.maven.project.MavenProject, org.a ...