在NCBI中下载SRA数据
目前,在NCBI中下载SRA数据主要有三种方式:
- 利用Aspera工具下载。
- 利用SRA Toolkit下载。
- 利用wget命令直接下载
第三种最为方便。其中的关键是得到下载数据的链接,即ftp的地址
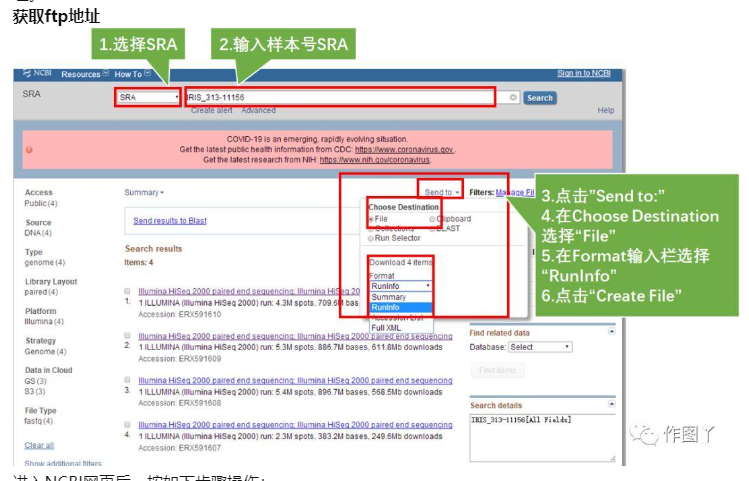
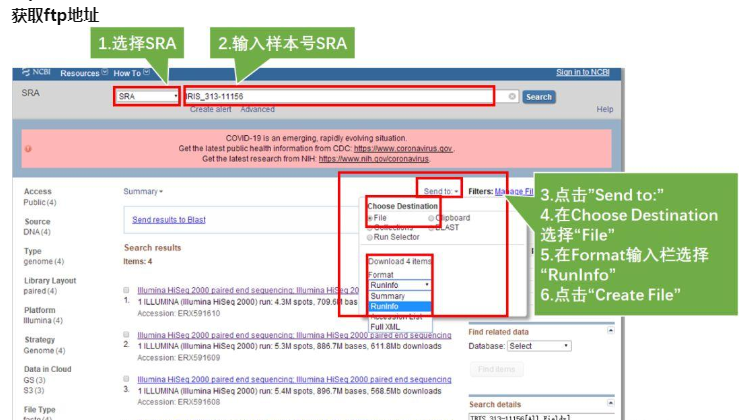
进入NCBI网页后,按如下步骤操作:
- Step1.设置NCBI的分类为:SRA
- Step2.输入感兴趣的样本号:IRIS_313-11156,点击Search,弹出四条item,说明该样本分四次run上级,我们需要全部下载
- Step3.点击右上角的Send to
- Step4.在Choose Destination中选择File
- Step5.在Format输入栏选择RunInfo
- Step6.点击Create File,此刻会生成一个名为SraRunInfo.csv的文件,图中标黄的一列即为不同次run数据的ftp地址。
wget -c 50 下载地址
若想批量下载则把下载地址放到一个list里面,然后运行下面的代码:
wget -c 50 -i list.txt
下面这个网址里面也有一些内容可供参考:
https://www.jianshu.com/p/0694fcb77157
https://www.cnblogs.com/zdwu/p/8473986.html
下载好的数据是sra压缩格式,这个格式是ncbi特有的一种格式,需要将此格式的文件转换成fastq文件的格式
sra是NCBI 推出的存储高通量数据的格式,而平常我们工作用得多是fastq格式。如果需要把sra 转成fastq,从
http://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?cmd=show&f=software&m=software&s=software
下载相应的软件。
或者下载最新的source code,在服务器上用make 编译。
然后使用如下命令行:
sra_sdk-2.0.0rc1/linux/rel/gcc/x86_64/bin/fastq-dump -A SRR034580 -D SRR034580.sra
这样就可以很简单的把sra格式转成fastq格式了。
转换 .sra 文件成 .fastq/fasta 文件
#single-end 单端测序
.../fastq-dump DRR000003.sra # 结果生成DRR000003.fastq
.../fastq-dump --fasta DRR000003.sra # 结果生成DRR000003.fastq
#pair-end 双端测序
.../fastq-dump --split-3 DRR002018.sra # 结果生成 DRR002018_1.fastq,DRR002018_2.fastq
REF:
http://blog.sina.com.cn/s/blog_4055a5940100o1mg.html
http://hi.baidu.com/wuyu466/item/152006eb4363eac3baf37d29
http://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software
http://blog.sina.com.cn/s/blog_70b2b6020100liee.html
在NCBI中下载SRA数据的更多相关文章
- NCBI下载sra数据(新)
今天要上NCBI下载sra数据发现没有下载的链接,网上查发现都是老的方法,NCBI页面已经变更,于是看了NCBI的help,并且记录下来新版的sra数据下载方法,要用NCBI的工具SRA Tool ...
- NCBI下载SRA数据
从NCBI下载数据本来是一件很简单的事情,但是今天碰到几个坑: 1.paper里没有提供SRA数据号.也没有提供路径: 2.不知道文件在ftp的地址,不能直接用wget下载 所以通过在NCBI官网,直 ...
- 用R包来下载sra数据
1)介绍 我们用SRAdb library来对SRA数据进行处理. SRAdb 可以更方便更快的接入 metadata associated with submission, 包括study, sa ...
- Windows系统中下载Earthdata数据
总的来说,为四大步: 1.注册Earthdata用户. 注册时需注意的是,最好把所有需打勾的都勾上,在最后[注册]按钮前,弹出[人机验证]才能注册成功.如果注册不成功,除了检查用户名和密码是否符合要求 ...
- 如何利用efetch从NCBI中批量下载数据?
目录 找序列 下序列 假设我要从NCBI中下载全部水稻的mRNA序列,如何实施? 找序列 第一步,肯定是找到相关序列. 我从ncbi taxonomy进入,搜索oryza.因为要搜索mRNA核酸序列, ...
- 2021-2-3-利用anaconda+prefetch+aspera从NCBI的SRA数据库中下载原始测序数据
目录 1.Conda连接不上镜像源问题 2. aspera不能再独立使用 3.使用prefetch搭配aspera 4. prefetch下载方法 记录下下载过程,为自己和后人避坑. 1.Conda连 ...
- NCBI SRA数据预处理
SRA数据的的处理流程大概如下 一.SRA数据下载. NCBI 上存储的数据现在大都存储为SRA格式. 下载以后就是以SRA为后缀名. 这里可以通过三种方式下载SRA格式的数据. 1.通过http方式 ...
- 在ASP.NET MVC中利用Aspose.cells 将查询出的数据导出为excel,并在浏览器中下载。
正题前的唠叨 本人是才出来工作不久的小白菜一颗,技术很一般,总是会有遇到一些很简单的问题却不知道怎么做,这些问题可能是之前解决过的.发现这个问题,想着提升一下自己的技术水平,将一些学的新的'好'东西记 ...
- 利用Aspose.cells 将查询出的数据导出为excel,并在浏览器中下载。
正题前的唠叨 本人是才出来工作不久的小白菜一颗,技术很一般,总是会有遇到一些很简单的问题却不知道怎么做,这些问题可能是之前解决过的.发现这个问题,想着提升一下自己的技术水平,将一些学的新的‘好’东西记 ...
- 2. 在TCGA中找到并下载意向数据
听说过别人用生信分析"空手套白狼"的故事吧想做吗好想学哦~ 或多或少都知道GEO和TCGA这些公共数据库吧!那么你知道怎么在数据库上找到意向数据,并且成功下载呢?这第一步要难倒一大 ...
随机推荐
- oralce sql 缓存查询及删除
--缓存查询语句 V$SQLAREA 视图记录sql 执行情况(加载次数/用时/Id....) 常用字段 ADDRESS:SQL语句在SGA中的地址. 这两列被用于鉴别SQL语句,有时,两条不同的语句 ...
- C# 两个list集合合并成一个,及升序降序
C# List集合合并 在开发过程中.数组和集合的处理是最让我们担心.一般会用for or foreach 来处理一些操作.这里介绍一些常用的集合跟数组的操作函数. 首先举例2个集合A,B. L ...
- BIP 两个请求成功后,才能做某一件事
//保存前校验 let SetXStatus = 0; viewModel.on("beforeSave", function (args) { let _this = this; ...
- 关于给widget添加属性
在django中,我们通过修改Form/ModelForm的初始化函数__init__修改表单的显示样式,其中修改widget的属性操作和字典操作一致. 1.给widget添加属性 说明:这是在不影响 ...
- vue高级进阶( 三 ) 组件高级用法及最佳实践
vue高级进阶( 三 ) 组件高级用法及最佳实践 世界上有太多孤独的人害怕先踏出第一步. ---绿皮书 书接上回,上篇介绍了vue组件通信比较有代表性的几种方法,本篇主要讲述一下组件的高级用法和最 ...
- 记录一次vue的报错
1.主要代码 <div class="hello-ezuikit-js" v-for="(item,index) in list"> <div ...
- R语言码农的Scala学习心得
为了MLib,最近是铁了心要学好Spark.关注本博客的朋友应该知道我的主力语言是R,无论是训练模型还是做Elasticsearch,都是通过R脚本来操作的.之前的<通过 Spark R 操作 ...
- 淘淘商城项目技术点-9:使用FTPClient及FtpUtil工具类将图片上传至ngnix图片服务器
package com.taotao.controller; import com.taotao.common.utils.FtpUtil; import org.apache.commons.net ...
- SAP 结构转JSON
*使用方式 jsonstr = zui2_json=>serialize( data = ls_in compress = abap_true pretty_name = zui2_json=& ...
- 2022.11.09 NOIP2022 模拟赛六
科学 Source:CF461C Appleman and a Sheet of Paper,*2200. 注意到对于 \(p\le \lfloor \frac {now}{2}\rfloor\),直 ...