FASTQ格式
FASQT格式是用于存储生物序列(通常是核苷酸序列)及其相应的碱基质量分数的一种文本格式。为简洁起见,序列字母和质量分数均使用单个ASCII字符进行编码。最初由Wellcome Trust Sanger Institute(桑格研究所)开发用于捆绑FASTA格式的序列和其碱基质量分数的,现在已成为存储Illumina Genome Analyzer(Illumina基因组分析仪)等高通量测序仪的标准输出格式。
FASTQ文件格式
- 第1行,以“@” 字符开头,后面跟着一个序列标识符和一个可选的描述(像FASTA格式标题行)。
- 第2行,原始序列字符。
- 第3行,以“+”字符开头,后面可选地跟着相同的序列标识符或者任何描述。
- 第4行,第2行序列每个碱基的质量编码值,包含与第2行字符数量相同的符号。
包含单个序列的FASTQ文件示例如下:
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
字符“!”和“~”分别代表最低和最高质量,下面是ASCII代表质量值从左到右顺序递增:
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~Illumina序列标识符
来自Illumina软件的序列使用系统标识符:
@HWUSI-EAS100R:6:73:941:1973#0/1
| HWUSI-EAS100R | the unique instrument name |
|---|---|
| 6 | flowcell lane |
| 73 | tile number within the flowcell lane |
| 941 | 'x'-coordinate of the cluster within the tile |
| 1973 | 'y'-coordinate of the cluster within the tile |
| #0 | index number for a multiplexed sample (0 for no indexing) |
| /1 | the member of a pair, /1 or /2 (paired-end or mate-pair reads only) |
Illumina流程1.4版本以后,使用#NNNNNN代替#0作为多重ID,NNNNNN是多重标签的序列。
@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG
| EAS139 | the unique instrument name |
|---|---|
| 136 | the run id |
| FC706VJ | the flowcell id |
| 2 | flowcell lane |
| 2104 | tile number within the flowcell lane |
| 15343 | 'x'-coordinate of the cluster within the tile |
| 197393 | 'y'-coordinate of the cluster within the tile |
| 1 | the member of a pair, 1 or 2 (paired-end or mate-pair reads only) |
| Y | Y if the read is filtered, N otherwise |
| 18 | 0 when none of the control bits are on, otherwise it is an even number |
| ATCACG | index sequence |
请注意,最新版本的Illumina软件会输出样本编号(从样本表中读取替代)代替索引序列。例如,以下情况可能出现在批次的第一个样本中:
@EAS139:136:FC706VJ:2:2104:15343:197393 1:N:18:1
NCBI SRA数据
来自NCBI/EBI SRA数据库的FASTQ文件通常包含如下一个描述:
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC
+SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC
在本例中,由一个NCBI分配的标识符、描述保存Solexa/Illumina的原始标识符和read长度。测序是在双端模式下进行的(~500bp插入大小),详见SRR001666。值得注意的是,当使用带缺省参数设置的fastq-dump从SRA数据中提取数据时,fastq-dump将正向read和反向read的序列连接在一起,提取的序列具有双倍长度,丢失了双端测序信息。
$ /opt/sratoolkit.2.5.7-centos_linux64/bin/fastq-dump SRR001666
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=72
GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACCAAGTTACCCTTAACAACTTAAGGGTTTTCAAATAGA
+SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=72
IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9ICIIIIIIIIIIIIIIIIIIIIDIIIIIII>IIIIII/
@SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=72
GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGAAGCAGAAGTCGATGATAATACGCGTCGTTTTATCAT
+SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=72
IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBIIIIIIIIIIIIIIIIIIIIIIIGII>IIIII-I)8I
更好的方法是保存原始材料并分成两个或三个文件(正向、反向、单个),例如:
$ /opt/sratoolkit.2.5.7-centos_linux64/bin/fastq-dump --origfmt --split-3 SRR001666
$ head SRR001666_1.fastq SRR001666_2.fastq
==> SRR001666_1.fastq <==
@071112_SLXA-EAS1_s_7:5:1:817:345
GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC
+071112_SLXA-EAS1_s_7:5:1:817:345
IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC
@071112_SLXA-EAS1_s_7:5:1:801:338
GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGA
+071112_SLXA-EAS1_s_7:5:1:801:338
IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBI ==> SRR001666_2.fastq <==
@071112_SLXA-EAS1_s_7:5:1:817:345
AAGTTACCCTTAACAACTTAAGGGTTTTCAAATAGA
+071112_SLXA-EAS1_s_7:5:1:817:345
IIIIIIIIIIIIIIIIIIIIDIIIIIII>IIIIII/
@071112_SLXA-EAS1_s_7:5:1:801:338
AGCAGAAGTCGATGATAATACGCGTCGTTTTATCAT
+071112_SLXA-EAS1_s_7:5:1:801:338
IIIIIIIIIIIIIIIIIIIIIIGII>IIIII-I)8I
另请注意,NCBI已将此FASTQ数据从原始Solexa/Illumina编码转换为Sanger标准(请参阅下面的编码)。
变动
质量
质量值Q是p的整数映射(即相应碱基判定不正确的概率),主要有两种不同的公式被使用。第一种是评估碱基判定的可靠性的不标准Sanger变体,也称为Phred质量分数:

Solexa流程(即与Illumina Genome Analyzer一起交付的软件)较早使用了不同的映射编码概率p/(1-p),而不是p:

虽然两个映射在更高质量值处渐近地相同,但它们在较低质量水平下不同(即,近似p> 0.05等同Q <13)。
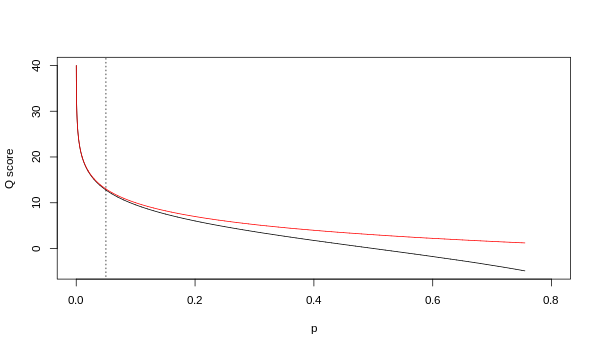
使用Sanger(红色)和Solexa(黑色)方程(如上所述)的Q和p之间的关系。 垂直虚线表示p = 0.05,或等同地Q≈13。
llumina流程1.4版用户指南(附录B,第122页)指出:“评分定义为:Q=10*log10(p/(1-p)),其中p是对应碱基判定的概率”。Illumina流程1.5版用户指南(第5页新增内容)对此进行了修改,“流程1.3版质量评分方案已更改为Phred评分方案,质量得分按Phred+64被转换为ASCII字符表示。”
编码
- Sanger格式可以使用ASCII 33~126字符表示0~93的Phred质量分数(尽管在原始read数据中,Phred质量分数很少超过60,但在组装或比对中可能有更高的分数)。这种方法也用于SAM格式中。根据seqanswers.com 上的公告,到2011年2月底,Illumina最新版本(1.8)的CASAVA流程将直接产生Sanger格式的fastq文件。
- Solexa/Illumina 1.0格式可以使用ASCII 59~126字符表示-5~62的Solexa/Illumina评分(原始read数据Solexa评分仅为-5~40)。
- 从Illumina 1.3版本开始,到Illumina 1.8版本,使用ASCII 64~126表示0~62的Phred质量分数(原始数据中Phred分数仅为0~40)。
- 从Illumina1.5版本开始,到Ilumina 1.8版本,Phred分数为0~2的含义略有不同。值0和1不在使用,值2(ASCII字符“B”)作为read片段结束的质量控制指示器。Illumina手册(第30页)内容:如果read以质量最差的一段(Q15或更低)结束,则片段中的所有质量值将被值2替换(ASCII字符“B”)…此Q2指标不能表示特定的错误率,而是表示read的特定最终部分不应用于进一步分析。此外,在Illumina 1.6版本之前,质量分数编码为“B”的碱基也可能在read内部发生,如下所示:
@HWI-EAS209_0006_FC706VJ:5:58:5894:21141#ATCACG/1
TTAATTGGTAAATAAATCTCCTAATAGCTTAGATNTTACCTTNNNNNNNNNNTAGTTTCTTGAGATTTGTTGGGGGAGACATTTTTGTGATTGCCTTGAT
+HWI-EAS209_0006_FC706VJ:5:58:5894:21141#ATCACG/1
efcfffffcfeefffcffffffddf`feed]`]_Ba_^__[YBBBBBBBBBBRTT\]][]dddd`ddd^dddadd^BBBBBBBBBBBBBBBBBBBBBBBB
- 对于原始的read,质量分数的范围取决于所使用的技术和碱基判定标准,但对于最新的Illumina流程,通常会达到41.由于最大观察质量分数以前仅为40,因此各种脚本工具在遇到质量值大于40的数据时会中断。而对于经过过处理的read,质量分数可能会更高。例如:Illumina的Long Read Sequencing Service(以前的Moleculo)中read的质量分数为45。
SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS.....................................................
..........................XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX......................
...............................IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII......................
.................................JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ.....................
LLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLL....................................................
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~
| | | | | |
33 59 64 73 104 126
0........................26...31.......40
-5....0........9.............................40
0........9.............................40
3.....9..............................41
0.2......................26...31........41 S - Sanger Phred+33, raw reads typically (0, 40)
X - Solexa Solexa+64, raw reads typically (-5, 40)
I - Illumina 1.3+ Phred+64, raw reads typically (0, 40)
J - Illumina 1.5+ Phred+64, raw reads typically (3, 41)
with 0=unused, 1=unused, 2=Read Segment Quality Control Indicator (bold)
(Note: See discussion above).
L - Illumina 1.8+ Phred+33, raw reads typically (0, 41)
色域
SOLiD数据,除了第一个位置,序列信息存储在色域中。质量值是Sanger格式的质量值,比对工具在质量值的首选版本上有所不同:其中一些包括主要和核苷酸的质量评分,而另一些则没有。序列read存档文件包含此质量分数。
模拟
压缩
质量分数占FASTQ格式所需磁盘空间(压缩前)的一般左右,因此压缩质量值可显著降低存储需求并加快测序数据的分析和传输。文献中提到的压缩方式主要无损和有损压缩。例如,有损压缩QualComp,无损压缩SCALCE、fastqz。
从Hiseq 2500开始,Illumina提供了将粗粒度质量输出到质量表的选项。分积分数直接从经验质量分数表中计算得出,该分数表与测序实验中所使用的硬件、软件和化学物质有关。
文件拓展名
FASTQ文件并没有标准的文件拓展名,但通常都是.fq或.fastq。
格式转换器
- Biopython version 1.51 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- EMBOSS version 6.1.0 patch 1 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- BioPerl version 1.6.1 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- BioRuby version 1.4.0 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- BioJava version 1.7.1 onwards (interconverts Sanger, Solexa and Illumina 1.3+)
- MAQ can convert from Solexa to Sanger (use this patch to support Illumina 1.3+ files).
- fastx_toolkit The included fastq_quality_converter program can convert Illumina to Sanger
命令行转换
FASTQ to FASTA format:
zcat input_file.fastq.gz | awk 'NR%4==1{printf ">%s\n", substr($0,2)}NR%4==2{print}' > output_file.fa
Illumina FASTQ 1.8 to 1.3
sed -e '4~4y/!"#$%&'\''()*+,-.\/0123456789:;<=>?@ABCDEFGHIJ/@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\\]^_`abcdefghi/' myfile.fastq # add -i to save the result to the same input file
Illumina FASTQ 1.3 to 1.8
sed -e '4~4y/@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\\]^_`abcdefghi/!"#$%&'\''()*+,-.\/0123456789:;<=>?@ABCDEFGHIJ/' myfile.fastq # add -i to save the result to the same input file
Illumina FASTQ 1.8 raw quality to binned quality (HiSeq Qtable 2.10.1, HiSeq 4000 )
sed -e '4~4y/!"#$%&'\''()*+,-.\/0123456789:;<=>?@ABCDEFGHIJKL/))))))))))----------77777<<<<<AAAAAFFFFFJJJJ/' myfile.fastq # add -i to save the result to the same input file
Illumina FASTQ 1.8 raw quality to clinto format (a visual block representation)
sed -e 'n;n;n;y/!"#$%&'\''()*+,-.\/0123456789:;<=>?@ABCDEFGHIJKL/▁▁▁▁▁▁▁▁▂▂▂▂▂▃▃▃▃▃▄▄▄▄▄▅▅▅▅▅▆▆▆▆▆▇▇▇▇▇██████/' myfile.fastq # add -i to save the result to the same input file
参考资料
转载请标明《http://www.cnblogs.com/yahengwang/p/8973948.html》
FASTQ格式的更多相关文章
- fasta/fastq格式解读
1)知识简介--------------------------------------------------------1.1)测序质量值 首先在了解fastq,fasta之前,了解一下什么是质量 ...
- 文件格式——fastq格式
fastQ格式 FASTQ是一种存储了生物序列(通常是核酸序列)以及相应的质量评价的文本格式. 他们都是以ASCII编码的.现在几乎是高通量测序的标准格式.NCBI Short Read Archiv ...
- Spark直接读入fastq格式的数据
输入文件: fastq格式 输出结果: kmer的频数和对应的kmer类型 系统环境Ubuntu单机版17.01 spark版本2.7 此次测试主要用到了RDD的函数foreach和zipWithIn ...
- 利用Bioperl的SeqIO模块解析fastq文件
测序数据中经常会接触到fastq格式的文件,比如说拿到fastq格式的原始数据后希望查看测序碱基的质量并去除低质量碱基.一般而言大家都是用现有的工具,比如说fastqc这个Java写的小程序,确实很好 ...
- 【转录组入门】3:了解fastq测序数据
操作:需要用安装好的sratoolkit把sra文件转换为fastq格式的测序文件,并且用fastqc软件测试测序文件的质量 作业:理解测序reads,GC含量,质量值,接头,index,fastqc ...
- 弗雷塞斯 从生物学到生物信息学到机器学习 转录组入门(3):了解fastq测序数据
sra文件转换为fastq格式 1 fastq-dump -h --split-3 也就是说如果SRA文件中只有一个文件,那么这个参数就会被忽略.如果原文件中有两个文件,那么它就会把成对的文件按*_1 ...
- sam格式详细说明
原文链接 https://www.jianshu.com/p/386f520e5de1 The SAM Format Specification(sam格式说明) 1 The SAM Format S ...
- 转录组入门(3):了解fastq测序数据
sra文件转换为fastq格式 fastq-dump -h --split-3 也就是说如果SRA文件中只有一个文件,那么这个参数就会被忽略.如果原文件中有两个文件,那么它就会把成对的文件按*_1.f ...
- C语言计算fastq文件GC含量
C语言小练习:计算非压缩fastq格式的GC含量 1 #include <stdio.h> 2 #include <stdlib.h> 3 #include <strin ...
随机推荐
- iOS开发常用第三方框架
1.网络通信 1.ASIHTTPRequest 这是一个经典的老库,功能完全而强大,但已经停止更新很久了(iOS5.0停止更新,但是我最近看github上这个项目有新改动).在不同iOS版本上略微有一 ...
- Django框架打印orm转换过程中的sql_模型层
LOGGING = { 'version': 1, 'disable_existing_loggers': False, 'handlers': { 'console':{ 'level':'DEBU ...
- Oozie-1-安装、配置 让Hadoop流动起来
版权声明:本文为博主原创文章,未经博主同意不得转载. https://blog.csdn.net/wl101yjx/article/details/27881739 写在前面一: 本文总结 基于Had ...
- webpack笔记1
1.设置多个入口起点 多用于提取公共类库 a.利用commonChunkPlugin const webpack= require('webpack'); const path = require(' ...
- Spring Boot2.0之 监控管理
Spring boot监控中心: 针对微服务的服务状态,服务器的内存变化(内存.线程.日志管理等)检测服务配置连接地址是否有用(有些懒加载的情况下,用的时候发现卧槽不能用)模拟访问,懒加载.统计有多少 ...
- 51nod 1737配对
题意:给定一个n个点的带边权树, 保证n是偶数,给这个树两两配对,使得配对后的点路径和最大,输出最大值. 其实是个很简单的题,但还是被绊了.这充分说明现在连简单题都做不来了555 单独考虑每条边.每 ...
- TCP/IP协议数据包文件PCAP分析器
一.设计原理 1.PCAP文件构成 参考http://blog.csdn.net/gulu_gulu_jp/article/details/50494909 PCAP文件由一个PCAP文件头和多个PC ...
- dead reckoning variation
Targeting A target is a structure of information that describes the state, and change of state, of a ...
- aop 例子(annotation方式实现)
面向切面编程(也叫面向方面),可以通过预编译方式和运行期动态代理实现在不修改源代码的情况下给程序动态统一添加功能的一种技术.AOP实际是GoF设计模式的延续,设计模式孜孜不倦追求的是调用者和被调用者之 ...
- django model中get()和filter()方法的区别
django的get()方法是从数据库的取得一个匹配的结果,返回一个对象,如果记录不存在的话,它会报错. django的filter()方法是从数据库的取得匹配的结果,返回一个对象列表,如果记录不存在 ...