R包对植物进行GO,KEGG注释
1、安装,加载所用到到R包
用BiocManager安装,可同时加载依赖包
source("https://bioconductor.org/biocLite.R")
BiocManager::install("clusterProfiler")
library(clusterProfiler) ##富集分析
library(topGO) ###画GO图
library(AnnotationHub) ##获取数据库
library(BiocFileCache) ##依赖包
library(dbplyr) ##依赖包
library(pathview) ##看KEGG pathway
2、利用annotataionHub去抓取目标orgDb
ah <- AnnotationHub() ##收索所有orgdb,到ah
unique(ah$dataprovider) ##可查看数据注释来源
query(ah, "Apis cerana") ##查找目标物种
tar_org <- ah[["AH62635"]] ##下载目标物种到org数据
3、了解org数据库
主要有5个函数
columns(x): 显示当前对象有哪些数据
keytypes(x): 有哪些keytypes可以用作select或keys的keytypes参数
keys(x, keytype, ...):返回当前数据对象的keys
select(x, keys, columns, keytype, ...):基于keys, columns和keytype以data.frame数据类型返回数据,可以是一对多的关系
mapIds(x, keys, column, keytype, ..., multiVals): 类似于select,只不过就返回一个列。3.1 以symbol形式展示
head(keys(tar_org,keytype = "SYMBOL"),30) ##默认为ENTREZID
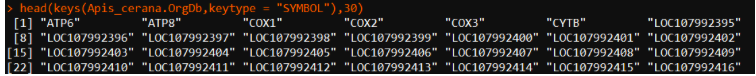
3.2、可以查看gene类型
keytypes(tar_org)
[1] "ARACYC" "ARACYCENZYME" "ENTREZID" "ENZYME" "EVIDENCE" "EVIDENCEALL" "GENENAME"
[8] "GO" "GOALL" "ONTOLOGY" "ONTOLOGYALL" "PATH" "PMID" "REFSEQ"
[15] "SYMBOL" "TAIR" 3.3、select则是根据你提供的key值去查找注释数据库,返回你需要的columns信息
> select(tar_org, keys= "AGO1", columns=c("TAIR","GO"),keytype = "SYMBOL")
'select()' returned 1:many mapping between keys and columns
SYMBOL TAIR GO EVIDENCE ONTOLOGY
1 AGO1 AT1G48410 GO:0004521 IDA MF
2 AGO1 AT1G48410 GO:0005515 IPI MF
3 AGO1 AT1G48410 GO:0005634 IDA CC
4 AGO1 AT1G48410 GO:0005737 IDA CC
5 AGO1 AT1G48410 GO:0005737 ISM CC
6 AGO1 AT1G48410 GO:0005737 TAS CC
7 AGO1 AT1G48410 GO:0005829 IDA CC
8 AGO1 AT1G48410 GO:0006306 RCA BP
9 AGO1 AT1G48410 GO:0006342 RCA BP
10 AGO1 AT1G48410 GO:0006346 RCA BP️找到差异基因后,必须得确定你得基因号是对应ENTREZID 或者SYMBOL,,是属于哪种类型,若没有符合上述类型,可自行找到NCBI上得数据名称,进行blast更换名字。
4、进行作图
统一将差异基因名字改为ENTREZID,防止在做GO分析的时候出现报错,需要将symbolID转换成ENTREZID:用mapIds函数就可以转换ID。
DEG.entrez_id = mapIds(x = tar_org, #### 数据库
keys = DEG.gene_symbol, #####差异基因名字
keytype = "SYMBOL", ####差异基因类型是SYMBOL
column = "ENTREZID") #####转换为ENTREZID
这时就已经把symbolID转换成ENTREZID了,但会出现个别的转换不成功的情况,就是图中NA的地方,我们进行以下操作即可去掉:
DEG.entrez_id = na.omit(DEG.entrez_id)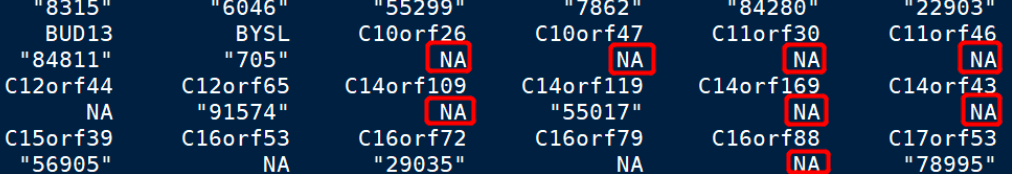
4.1、GO分析代码
BP(Biological process)层面上的富集分析:
erich.go.BP = enrichGO(gene = DEG.entrez_id, ###差异基因ID
OrgDb = tar_org, ###数据库
keyType = "ENTREZID", ##基因ID类型
ont = "BP", ##对BP进行GO分析
pvalueCutoff = 0.5, ###fisher检验对p值
qvalueCutoff = 0.5) ###对p值进行校对对q值,一般大于p值
作图:
dotplot(erich.go.BP)解读BP层面富集分析图:
横坐标是GeneRatio,意思是说输入进去的基因,它每个term(纵坐标)站整体基因的百分之多少。圆圈的大小代表基因的多少,图中给出了最大的圆圈代表60个基因,圆圈的颜色代表P-value,也就是说P-value越小gene count圈越大,这事就越可信。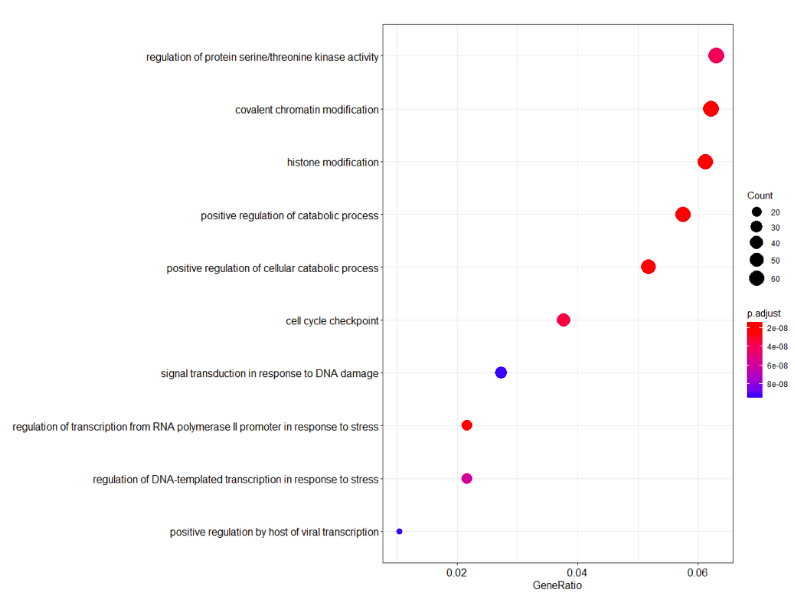
也可以画柱形图
barplot(erich.go.CC)
一般GO分析画这两个图就可以了,有时也把GO分析画成树形图,可以更加帮助我们理解。
plotGOgraph(erich.go.BP)树状图很大,所以我们用代码把它存成pdf,学习下如何用代码
pdf(file="./enrich.go.bp.tree.pdf",width = 10,height = 15)
plotGOgraph(erich.go.BP)
dev.off()
至此,GO分析就做完了 ----> over
4.2、KEGG pathway 介绍
KEGG pathway是最常用的功能注释数据库之一,可以利用KEGG 的API获取一个物种所有基因对应的pathway注释,human对应的API 链接如下
http://rest.kegg.jp/link/hsa/pathway 人类
http://rest.kegg.jp/link/soe/pathway 菠菜
path:hsa00010 hsa:10327
path:hsa00010 hsa:124
path:hsa00010 hsa:125
第一列为pathway编号,第二列为基因编号。这里只提供了pathway编号,我们还需要pathway对应的描述信息,同样也可以通过以下API链接得到
http://rest.kegg.jp/list/
通过该链接可以获得如下内容
path:map00010 Glycolysis / Gluconeogenesis
path:map00020 Citrate cycle (TCA cycle)
path:map00030 Pentose phosphate pathway
path:map00040 Pentose and glucuronate interconversions
path:map00051 Fructose and mannose metabolism
第一列为pathway编号,第二列为具体的描述信息。需要注意的是,pathway是一个跨物种的概念,原始的pathway编号为map或者ko加数字,对于特定物种,改成物种对应的三字母缩写, 比如human对应hsa, 所有拥有pathway信息的物种和对应的三字母缩写见如下链接
https://www.genome.jp/kegg/catalog/org_list.html
clusterProfiler也是通过KEGG API去获取物种对应的pathway注释,对于已有pathway注释的物种,我们只需要知道对应的三字母缩写, clusterProfiler就会联网自动获取该物种的pathway注释信息。
和GO富集分析类似,对于KEGG的富集分析也包含以下两种
过表征分析 (over representation analysis, ORA) ###先会筛选,并挑选出我们感兴趣对基因
基因富集分析 (gene set enrichment analysis, GSEA) ###不进行筛选
enrich.KEGG.BP <- enrichKEGG(gene = test_sample, #### 差异基因ID ENTREZID
keyType = "kegg", ####key类型
organism = "soe", ###物种3字母
pvalueCutoff = 0.05,
pAdjustMethod = "BH",
qvalueCutoff = 0.1,)
4.2.1、柱状图
barplot(enrich.KEGG.BP, showCategory = 10)
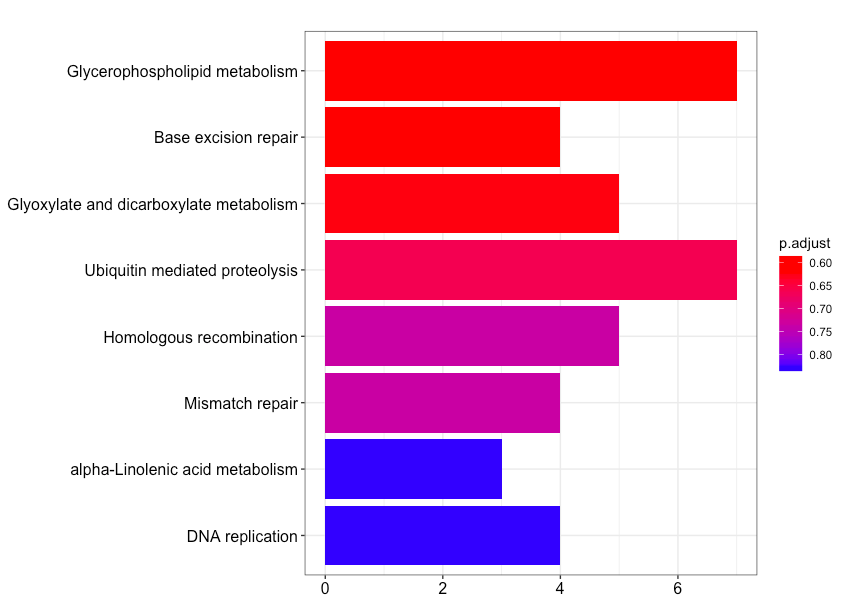
横轴为该pathway的差异基因个数,纵轴为富集到的pathway的描述信息, showCategory指定展示的pathway的个数,默认展示显著富集的top10个,即p.adjust最小的10个。注意的颜色对应p.adjust值,从小到大,对应蓝色到红色。
4.2.2、点图
dotplot(enrich.KEGG.BP, showCategory = 10)
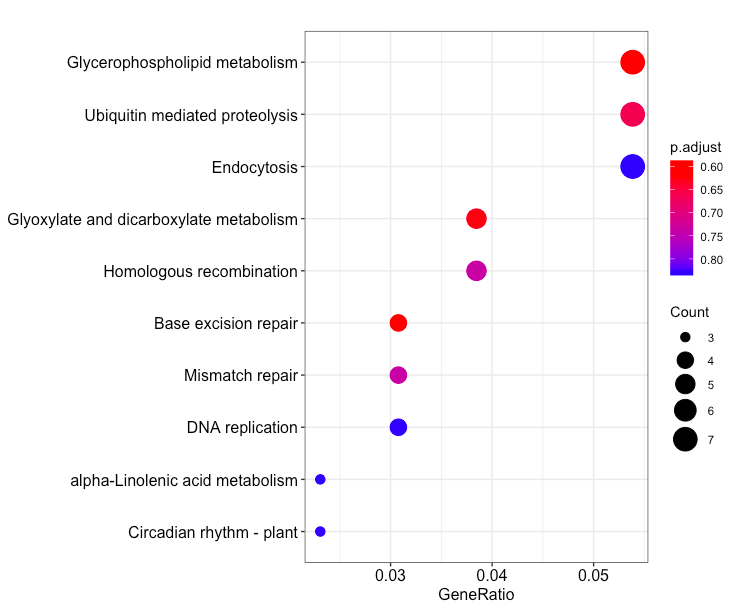
横轴为GeneRatio, 代表该pathway下的差异基因个数占差异基因总数的比例,纵轴为富集到的pathway的描述信息, showCategory指定展示的pathway的个数,默认展示显著富集的top10个,即p.adjust最小的10个。图中点的颜色对应p.adjust的值,从小到大,对应蓝色到红色,大小对应该GO terms下的差异基因个数,个数越多,点越大。
4.2.3、Gene-Concept Network
前面的 两款神器 两个函数,都只能展示富集最显著的 GO term,而函数 cnetplot() 可以将基因与生物学概念 (e.g.* GO terms or KEGG pathways) 的关系绘制成网状图。对于基因和富集的pathways之间的对应关系进行展示,如果一个基因位于一个pathway下,则将该基因与pathway连线,用法如下
cnetplot(enrich.KEGG.BP, showCategory = 5)
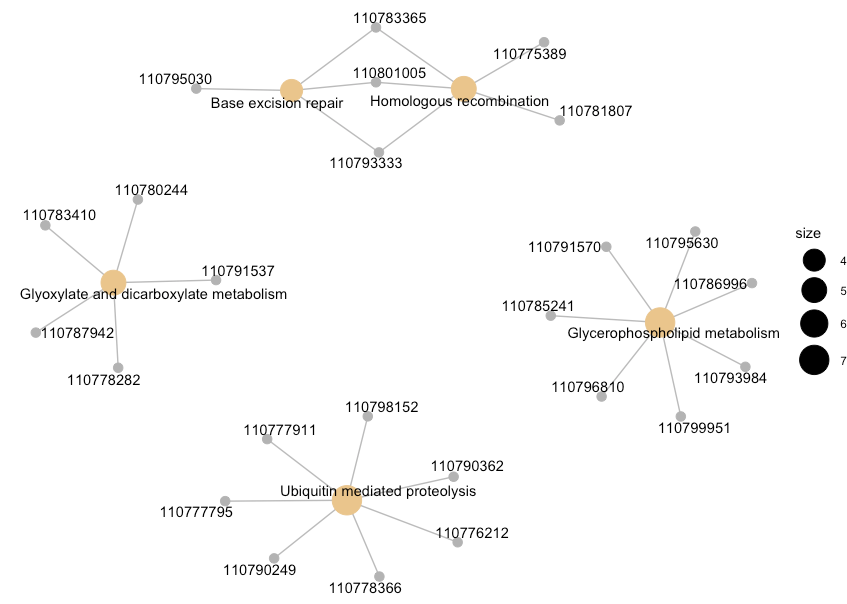
图中灰色的点代表基因,黄色的点代表富集到的pathways, 默认画top5富集到的pathwayss, pathways节点的大小对应富集到的基因个数。数字就是基因ID,如果需要更换,可以更换keytype,或者直接在enrich.KEGG.BP 的结果中进行相同ID更换
cnetplot(enrich.KEGG.BP,circular=T, ###画为圈图 colorEdge=T) ##线条用颜色区分
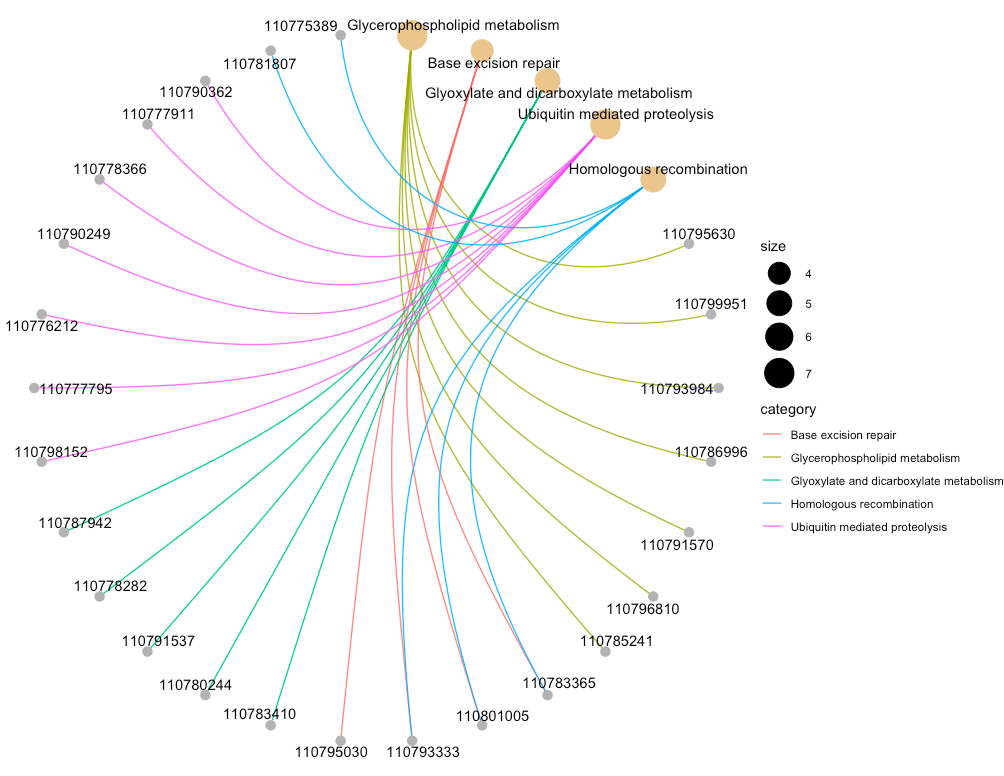
4.2.4、Enrichment Map
Enrichment Map 可以将富集条目和重叠的基因集整合为一个网络图,相互重叠的基因集则趋向于成簇,从而易于分辨功能模型。对于富集到的pathways之间的基因重叠关系进行展示,如果两个pathway的差异基因存在重叠,说明这两个节点存在overlap关系,在图中用线条连接起来,用法如下
emapplot(enrich.KEGG.BP)
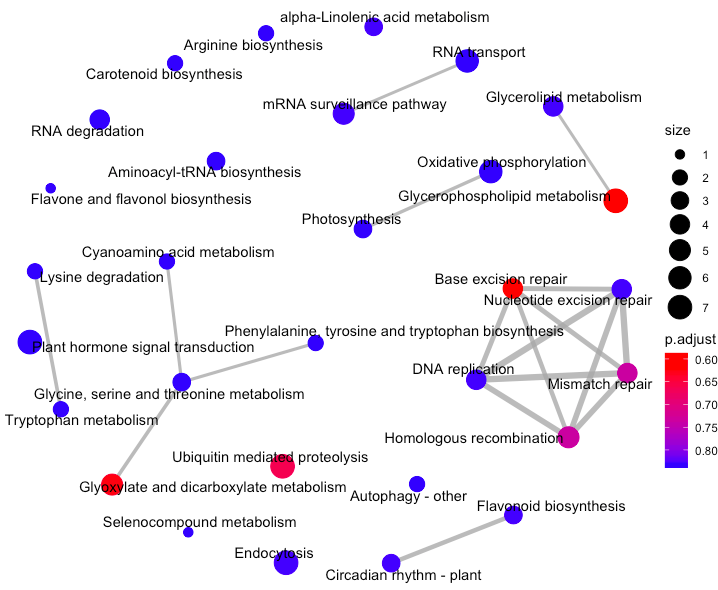
每个节点是一个富集到的pathway, 默认画top30个富集到的pathways, 节点大小对应该pathway下富集到的差异基因个数,节点的颜色对应p.adjust的值,从小到大,对应蓝色到红色。
4.2.5 browseKEGG
browseKEGG(enrich.KEGG.BP,"soe00564") ###画出某一特定pathway的图
关于KEGG解释,可以查看链接
https://www.jianshu.com/p/f90ed1c52079
4.2.6 pathview 包里的 上帝视角 PATHVIEW!
pathview(gene.data = test_sample, ##是需要提供的基因向量,默认是Entrez_ID。其由gene.idtype决定
pathway.id = "soe00564", ###指的是在KEGG中的ID
species = "soe",
kegg.native = TRUE,###默认是TRUE输出完整pathway的png格式文件,反之输出仅是输入的基因列表的pdf文件。
)
感觉结果和 browseKEGG 差不多
持续整理。。。
关注下方公众号可获得更多精彩

参考:https://www.jianshu.com/p/ae94178918bc GO
https://www.jianshu.com/p/47b5ea646932?utm_source=desktop&utm_medium=timeline GO
https://www.cnblogs.com/djx571/p/10271874.html GO
https://blog.csdn.net/weixin_43569478/article/details/83744384 KEGG
https://www.jianshu.com/p/f90ed1c52079 KEGG
https://www.jianshu.com/p/e133ab3169fa KEGG
R包对植物进行GO,KEGG注释的更多相关文章
- R 包
[下面列出每个步骤最有用的一些R包] .数据导入 以下R包主要用于数据导入和保存数据: feather:一种快速,轻量级的文件格式:在R和python上都可使用 readr:实现表格数据的快速导入 r ...
- 极简 R 包建立方法--转载
https://cosx.org/2013/11/building-r-packages-easily/ 最近想试一下捣腾一个 R 包出来,故参考了一些教程.现在看到的最好的就是谢益辉大大之前写过的开 ...
- R语言无网络安装R包,彻底解决依赖问题!
R version: 3.5.3, 3.6.3 更新日期: 2020-9-10 大家测试后多提建议哈, 有问题我会持续更新的 在工作中,我们使用的服务器通常是不能联外网的,这在安装R包的时候产生了巨大 ...
- 标准化R包开发流程
3个武器: devtools:各种开发小工具的合集,让开发变得简单,非常实用 roxygens:通过注释的方式,生成文档,远离LaTex的烦恼 testthat:单元测试,让R包稳定.健壮.减少升级的 ...
- R包MetaboAnalystR安装指南(Linux环境非root)
前言 这是代谢组学数据分析的一个R包,包括用于代谢组学数据分析.可视化和功能注释等众多功能.最近有同事在集群中搭建蛋白和代谢流程,安装这个包出现了问题,于是我折腾了一上午. 这个包的介绍在:https ...
- R包开发过程记录
目的 走一遍R包开发过程,并发布到Github上使用. 步骤 1. 创建R包框架 Rsutdio --> File--> New Project--> New Directory - ...
- 普通用户安装 R 包
转自 http://bnuzhutao.cn/archives/901 一般 R 语言的书籍上,介绍安装 R 包的方法都是这样的: install.packages("packagename ...
- R包介绍
R语言的使用,很大程度上是借助各种各样的R包的辅助,从某种程度上讲,R包就是针对于R的插件,不同的插件满足不同的需求,截至2013年3月6日,CRAN已经收录了各类包4338个. 一. R语言包的安装 ...
- R(三): R包原理及安装
包(package)是多个函数的集合,常作为分享代码的基本单元,代码封装成包可以方便其他用户使用.越来越多的R包正在由世界上不同的人所创建并分发,这些分发的R包,可以从CRAN 或 github 上获 ...
随机推荐
- Java/JDK/J2SE
Java8与JDK1.8与JDK8与J2SE8与J2SE1.8的区别是什么? Java是面向对象的编程语言,在我们开发Java应用的程序员的专业术语里,Java这个单词其实指的是Java开发工具,也就 ...
- 第五课第四周笔记2:Self-Attention 自注意力
Self-Attention 自注意力 让我们跳进去谈谈transformer的self-attention机制.如果您能了解本视频背后的主要思想,您就会了解变压器网络工作背后最重要的核心思想. 让我 ...
- [敏捷软工团队博客]Beta阶段发布声明
项目 内容 2020春季计算机学院软件工程(罗杰 任健) 博客园班级博客 作业要求 Beta阶段发布声明 我们在这个课程的目标是 在团队合作中锻炼自己 这个作业在哪个具体方面帮助我们实现目标 对Bet ...
- 第三次Alpha Scrum Meeting
本次会议为Alpha阶段第三次Scrum Meeting会议 会议概要 会议时间:2021年4月26日 会议地点:线上会议 会议时长:20min 会议内容简介:本次会议主要由每个人展示自己目前完成的工 ...
- NGINX杂谈——flask_limiter的IP获取(怎么拿到真实的客户端IP)
本篇博客将 flask_limiter 作为切入点,来记录一下自己对 remote_addr 和 proxy_add_x_forwarded_for 两个变量.X-Real-IP 和 X-Forwar ...
- 带你用AVPlayer实现音频和视频播放
项目概述 以下项目是基于AVPlayer的实际运用,实现音频播放.横竖屏视频切换播放.类似抖音的竖屏全屏播放效果. 项目地址:AVPlayerAudioVideo 如果文章和项目对你有帮助,还请给个S ...
- USART 硬件流控
流控的概念源于 RS232 这个标准,在 RS232 标准里面包含了串口.流控的定义.大家一定了解,RS232 中的"RS"是Recommend Standard 的缩写,即&qu ...
- 决策树 机器学习,西瓜书p80 表4.2 使用信息增益生成决策树及后剪枝
使用信息增益构造决策树,完成后剪枝 目录 使用信息增益构造决策树,完成后剪枝 1 构造决策树 1 根结点的选择 色泽 信息增益 根蒂 信息增益 敲声 信息增益 纹理 信息增益 脐部 信息增益 触感 信 ...
- 【JavaScript基础】Js的定时器(你想看的原理也在哟)
[JavaScript基础]Js的定时器(你想看的原理也在哟) 博客说明 文章所涉及的资料来自互联网整理和个人总结,意在于个人学习和经验汇总,如有什么地方侵权,请联系本人删除,谢谢! 说明 本章是经历 ...
- Luogu P1023 [NOIp2000提高组]税收与补贴问题 | 数学
题目链接 思路:列不等式组,然后解出不等式,得出答案的取值范围,最后取一个绝对值最小的答案就行了. #include<iostream> #include<cstdio> #i ...