kegg富集分析之:KEGGREST包(9大功能)
这个包依赖极有可能是这个:https://www.kegg.jp/kegg/docs/keggapi.html ,如果可以看懂会很好理解
由于KEGG数据库分享数据的策略改变,因此KEGG.db包不在能用,推荐KEGGREST包
But a number of years ago,KEGG changed their policy about sharing their data and so the KEGG.db package is no longer allowed to be current. Users who are interested in a more current pathway data are encouraged to look at the KEGGREST or reactome.db packages.
1、安装
if("KEGGREST" %in% rownames(installed.packages()) == FALSE) {source("http://bioconductor.org/biocLite.R");biocLite("KEGGREST")}
suppressMessages(library(KEGGREST))
ls('package:KEGGREST')
2、所有的对象及功能
keggConv:Convert KEGG identifiers to/from outside identifiers(ID转换功能)
keggFind:Finds entries with matching query keywords or other query data in a given database(即搜索功能)
keggGet:Retrieves given database entries(获取序列,图片等功能)
keggInfo:Displays the current statistics of a given database(即统计功能)
keggLink:Find related entries by using database cross-references(交互功能).
keggList:Returns a list of entry identifiers and associated definition for a given database or a given set of database entries.
listDatabases:Lists the KEGG databases which may be searched.
mark.pathway.by.objects:Client-side interface to obtain an url for a KEGG pathway diagram with a given set of genes marked(标记功能)
3、每个对象的功能简单使用
3.1、keggConv(Convert KEGG identifiers to/from outside identifiers,即ID转换功能)
语法:keggConv(target, source, querySize = 100)
例子:
## conversion from NCBI GeneID to KEGG ID for E. coli genes##
head(keggConv("eco", "ncbi-geneid"),2)
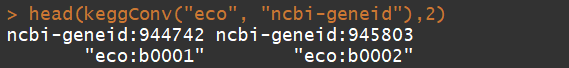
head(keggConv("ncbi-geneid", "eco"),2)

########conversion from KEGG ID to NCBI GI##########
head(keggConv("ncbi-proteinid", c("hsa:10458", "ece:Z5100")),2)

3.2)keggFind(Finds entries with matching query keywords or other query data in a given database,即检索功能)
语法:keggFind(database, query, option = c("formula", "exact_mass", "mol_weight"))
例子:
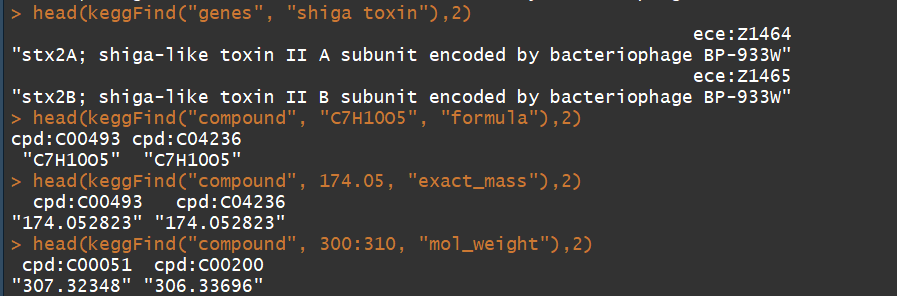
keggFind("genes", c("shiga", "toxin")) ## for keywords "shiga" and "toxin"
keggFind("genes", "shiga toxin") ## for keywords "shiga toxin"
keggFind("compound", "C7H10O5", "formula") ## for chemical formula "C7H10O5"
keggFind("compound", 174.05, "exact_mass") ## for 174.045 =< exact mass < 174.055
keggFind("compound", 300:310, "mol_weight") ## molecular weight 300 =< 310
3.3)keggGet (Retrieves given database entries,获取序列,图片等功能)
语法:keggGet(dbentries, option = c("aaseq", "ntseq", "mol", "kcf","image", "kgml"))
keggGet(c("cpd:C01290", "gl:G00092")) ## retrieves a compound entry and a glycan entry
keggGet(c("C01290", "G00092")) ## same as above, without prefixes
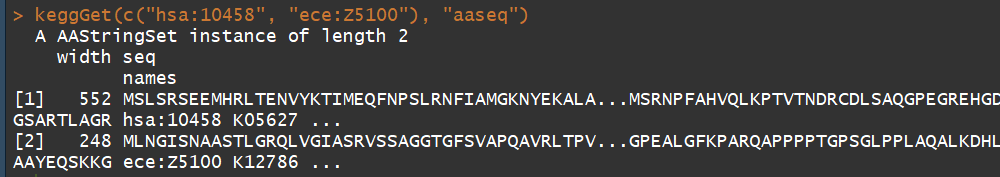
keggGet(c("hsa:10458", "ece:Z5100")) ## 检索 a human gene entry and an E.coli O157 gene entry
keggGet(c("hsa:10458", "ece:Z5100"), "aaseq") ## 检索 amino acid sequences of a human gene and an E.coli O157 gene
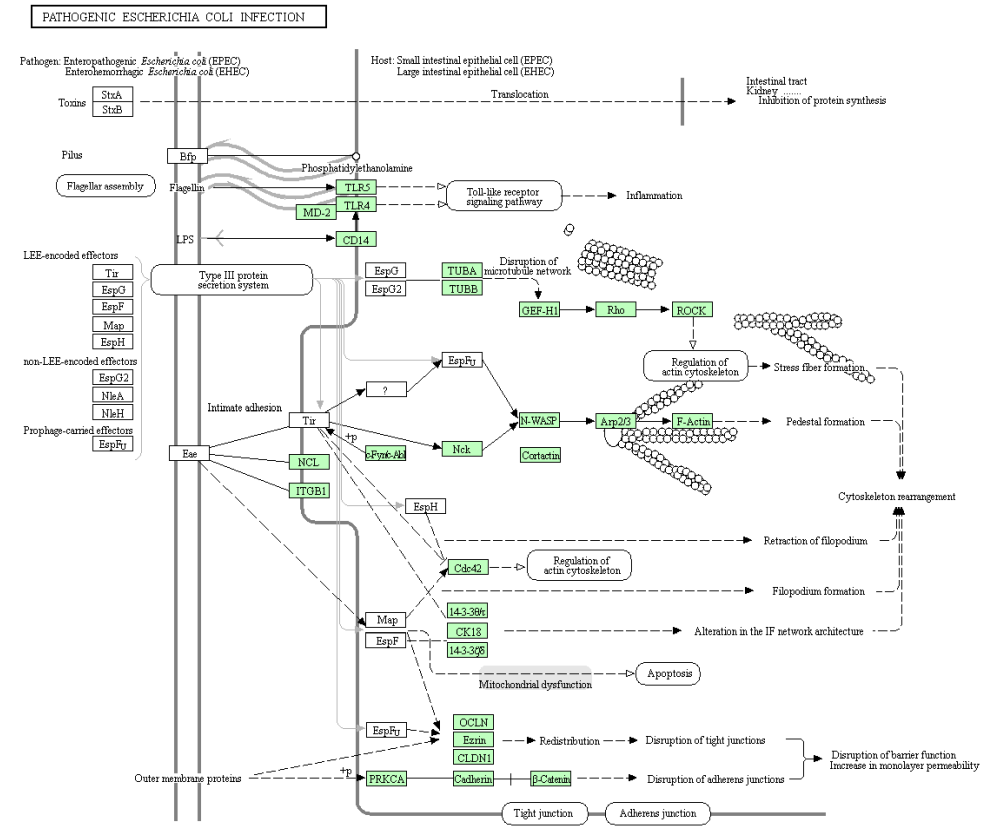
png <- keggGet("hsa05130", "image") ## retrieves the image file of a pathway map
t <- 'hsa05130.png'
library(png)
writePNG(png, t)
keggGet("hsa05130", "kgml")
hsa05130.png
3.4)keggInfo(Displays the current statistics of a given database,即统计功能)
语法:keggInfo(database)
head(keggInfo("kegg") ## displays the current statistics of the KEGG database
keggInfo("pathway") ## displays the number pathway entries including both the reference and organism-specific pathways
keggInfo("hsa") ##### displays the number of gene entries for the KEGG organism Homo sapiens
3.5)keggLink(keggLink Find related entries by using database cross-references.交互功能)
语法:keggLink(target, source)
head(keggLink("pathway", "hsa"),5) ### KEGG pathways linked from each of the human genes
head(keggLink("hsa", "pathway"),5) ## human genes linked from each of the KEGG pathways
head(keggLink("pathway", c("hsa:10458", "ece:Z5100")),5) ## KEGG pathways linked from a human gene and an E. coli O157 gene
head(keggLink("hsa:126"),5)

3.6) keggList (Returns a list of entry identifiers and associated definition for a given database or set of database entries,返回信息表)
用法:keggList(database, organism)
keggList("pathway") ## returns the list of reference pathways
keggList("pathway", "hsa") ## returns the list of human pathways
keggList("organism") ## returns the list of KEGG organisms with taxonomic classification
keggList("hsa") ## returns the entire list of human genes
keggList("T01001") ## same as above
keggList(c("hsa:10458", "ece:Z5100")) ## returns the list of a human gene and an E.coli O157 gene
keggList(c("cpd:C01290","gl:G00092")) ## returns the list of a compound entry and a glycan entry
keggList(c("C01290+G00092")) ## same as above (prefixes are not necessary)
3.7) listDatabases(Lists the KEGG databases which may be searched.查看有数据库)
用法:listDatabases()
例子:
listDatabases() ##查看有哪些数据库
3.8)mark.pathway.by.objects:Client-side interface to obtain an url for a KEGG pathway diagram with a given set of genes marked(标记功能,例如上调y用红框,下调用绿框)
语法:
mark.pathway.by.objects(pathway.id, object.id.list)
color.pathway.by.objects(pathway.id, object.id.list,fg.color.list, bg.color.list)
例子:
url <- mark.pathway.by.objects("path:eco00260",c("eco:b0002", "eco:c00263"))
if(interactive()){browseURL(url)}
url <- color.pathway.by.objects("path:eco00260",c("eco:b0002", "eco:c00263"),c("#ff0000", "#00ff00"), c("#ffff00", "yellow"))
kegg富集分析之:KEGGREST包(9大功能)的更多相关文章
- 【R】clusterProfiler的GO/KEGG富集分析用法小结
前言 关于clusterProfiler这个R包就不介绍了,网红教授宣传得很成功,功能也比较强大,主要是做GO和KEGG的功能富集及其可视化.简单总结下用法,以后用时可直接找来用. 首先考虑一个问题: ...
- KEGG富集分析散点图.md
输入数据格式 pathway = read.table("kegg.result",header=T,sep="\t") pp = ggplot(pathway ...
- DAVID 进行 GO/KEGG 功能富集分析
何为功能富集分析? 功能富集分析是将基因或者蛋白列表分成多个部分,即将一堆基因进行分类,而这里的分类标准往往是按照基因的功能来限定的.换句话说,就是把一个基因列表中,具有相似功能的基因放到一起,并和生 ...
- python scipy包进行GO富集分析p值计算
最近总是有需要单独对某一个类型的通路进行超几何分布的p值计算,这里记录一下python包的计算方法 使用scipy的stat里面的hypergeom.sf方法进行富集分析的p值计算 hsaxxxxx ...
- 富集分析DAVID、Metascape、Enrichr、ClueGO
前言 一般我们挑出一堆感兴趣的基因想临时看看它们的功能,需要做个富集分析.虽然公司买了最新版的数据库,如KEGG,但在集群跑下来嫌麻烦.这时网页在线或者本地化工具派上用场了. DAVID DAVID地 ...
- R: 修改镜像、bioconductor安装及go基因富集分析
1.安装bioconductor及go分析涉及的相关包 source("http://bioconductor.org/biocLite.R") options(BioC_mirr ...
- GO富集分析示例【华为云技术分享】
版权声明:本文为博主原创文章,遵循CC 4.0 BY-SA版权协议,转载请附上原文出处链接和本声明. 本文链接:https://blog.csdn.net/devcloud/article/detai ...
- DEPICT实现基因优化(gene prioritization)、gene set富集分析(geneset enrichment)、组织富集分析(tissue enrichment)
全基因组关联分析除了找到显著的关联位点,我们还可以做基因优化.geneset富集分析.组织富集分析,下面具体讲一讲怎么利用GWAS的summary数据做这个分析. summary数据就是关联分析的结果 ...
- GO富集分析 信号通路
基因富集分析是分析基因表达信息的一种方法,富集是指将基因按照先验知识,也就是基因组注释信息进行分类. 信号通路是指能将细胞外的分子信号经细胞膜传入细胞内发挥效应的一系列酶促反应通路.这些细胞外的分子信 ...
随机推荐
- 1、搭建HBase完全分布式集群
搭建完全分布式集群 HBase集群建立在hadoop集群基础之上,所以在搭建HBase集群之前需要把Hadoop集群搭建起来,并且要考虑二者的兼容性.现在就以5台机器为例,搭建一个简单的集群. 软件版 ...
- Django QueryDict
QueryDict默认是不可变的,同过将QueryDict对象的_mutable 属性的值设置成True就可以为其赋值.QueryDict对象的urlencode()方法将QueryDict转换为字符 ...
- Extjs Column布局常见问题及解决方法
原文地址:http://blog.csdn.net/weoln/article/details/4339533 第一次用Extjs的column布局时遇见了很多问题,记录下来,供大家参考.column ...
- Redis高速内存缓冲平台可视化监控之RedisLive配置实战
一.引用 这两天在弄Reids高速缓存平台的图形化监控,由于对于Python并不是很熟悉,安装过程中遇到了不少问题,包括: 1.python必备安装包的安装问题 2.Redis Live界面显示问题 ...
- Spring Boot + Jpa(Hibernate) 架构基本配置
本文转载自:https://blog.csdn.net/javahighness/article/details/53055149 1.基于springboot-1.4.0.RELEASE版本测试 2 ...
- wkhtmltopdf+itext实现html生成pdf文件的打印下载(适用于linux及windows)
目中遇到个根据html转Java的功能,在java中我们itext可以快速的实现pdf打印下载的功能,在itext中我们一般有以下三中方式实现 配置pdf模板,通过Adobe Acrobat 来设置域 ...
- [转][SVN]常用操作
1. Commit 提交当前代码到 SVN 服务器. 2. 引用第三方类库时,不要从安装位置引用,而是在解决方案下,添加一个 lib 的目录,把需要的程序集复制到这里,然后从 lib 目录引用. 3 ...
- 关于模板该不该用css强制编辑器文本开头空两格
关于模板该不该用css强制编辑器文本开头空两格这个问题,我很早之前就想说了,写惯了qq日志的童鞋都知道,qq空间的编辑器没有任何css控制,行头空两格是由用户自己控制,我写起日志又像流水账,长长的一篇 ...
- python 文本或句子切割,并保留分隔符
网上找了好久,都没有理想的解决方法.主要思想,利用正则表达式re.split() 分割,同时利用re.findall() 查找分隔符,而后将二者链接即可. # coding: utf- import ...
- keras的Embedding层
keras.layers.embeddings.Embedding(input_dim, output_dim, embeddings_initializer='uniform', embedding ...