gff/gtf格式
1)gff3及gtf2简介
一个物种的基因组测序完成后,需要对这些数据进行解读,首先要先找到这些序列中转录起始位点、基因、外显子、内含子等组成元件在染色体中的位置信息(即注释)后才能再进行深入的分析。gff/gtf是贮存这些注释信息的两种文件格式。
GFF(general feature format):这种格式主要是用来注释基因组。 现大部分利用的是第三版,即gff3。
GTF(gene transfer format):主要是用来对基因进行注释。当前所广泛使用的gtf格式为第二版,即gtf2 。
1.1)GFF3
GFF3允许使用#作为注释符号 ,除去注释外,主体部分共有9列。 GFF3中每一列的含义:seqid source type start end score strand strand attributes
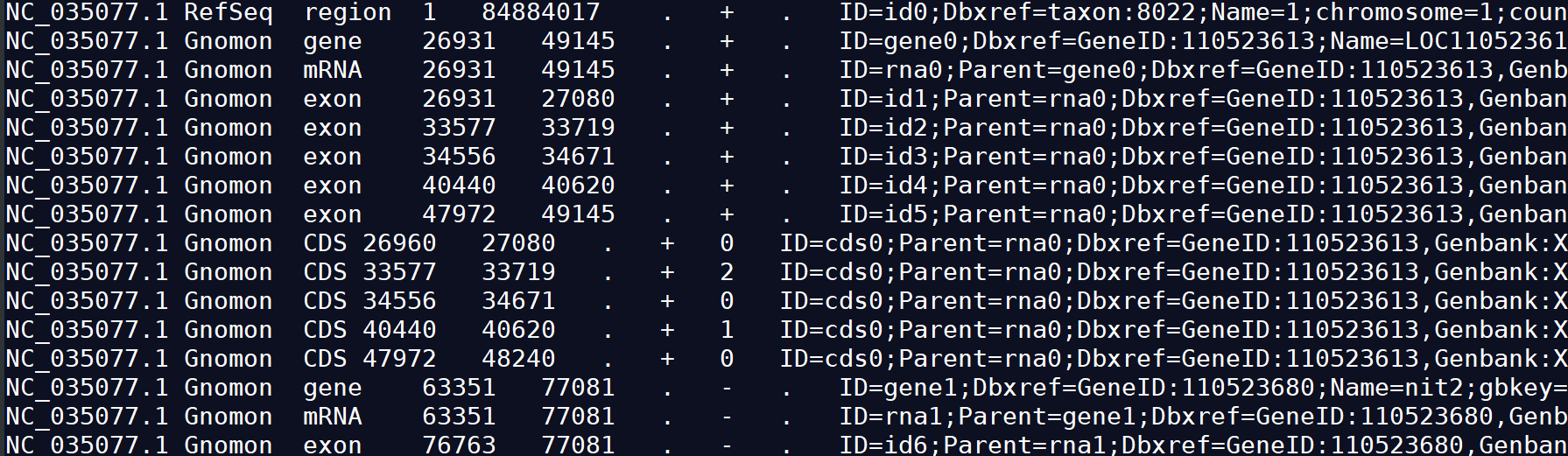
1) seqid :序列的id。(The name of the sequence where the feature is located.)
2)source:注释的来源,一般指明产生此gff3文件的软件或方法(e.g. Augustus or RepeatMasker)。如果未知,则用点(.)代替。
3)type: 类型,此处不受约束,但为下游分析方便,建议使用gene,repeat_region,exon,CDS,或SO对应编号等。
4)start:起始位置,从1开始计数(区别于bed文件从0开始计数)。
5)end:终止位置。
6)score:得分,注释信息可能性说明,可以是序列相似性比对时的E-values值或者基因预测是的P-values值。”.”表示为空。(indicates the confidence of the source on the annotated feature)
7)strand:“+”表示正链,“-”表示负链,“.”表示不需要指定正负链,“?” 表示未知.
8)phase :步进。仅对编码蛋白质的CDS有效,本列指定下一个密码子开始的位置。可以是0、1或2,表示到达下一个密码子需要跳过碱基个数。
9)attributes:属性。一个包含众多属性的列表,格式为“标签=值”(tag=value),不同属性之间以分号相隔。
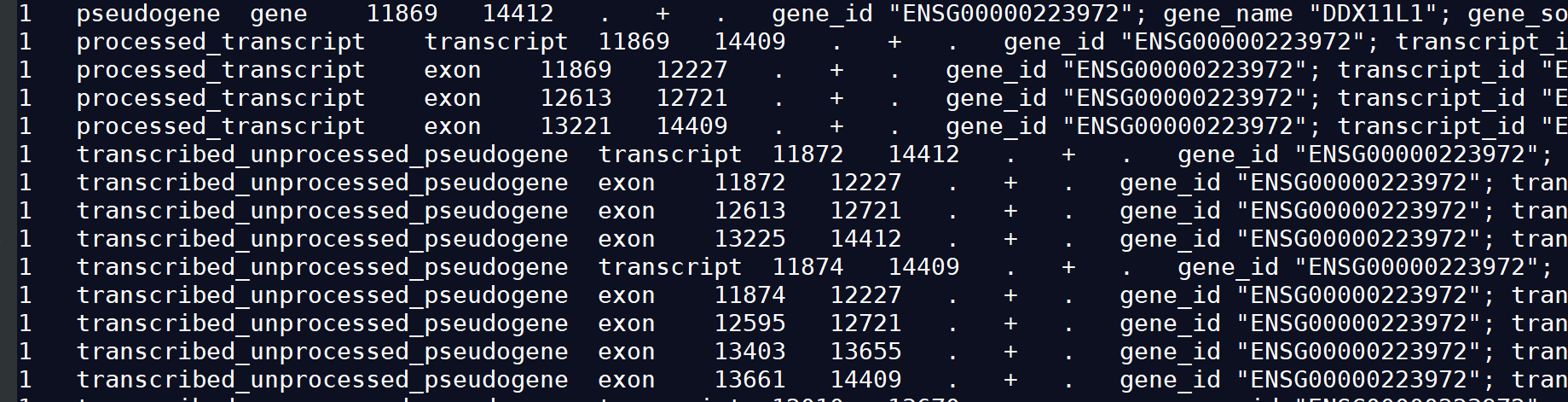
1.2)GTF2
gtf文件也是由9列组成,其中每一列含义:seqname source feature start end score strand frame attributes
1) seqname: 序列的名字。通常格式染色体ID或是contig ID。
2) source:注释的来源。通常是预测软件名或是公共数据库。
3) start:起始位置,从1开始计数。
4) end:终止位置。
5) feature :基因结构.根据所使用软件不同,feature types必须注明。CDS,start_codon,stop_codon是一定要含有的类型。
6) score :这一列的值表示对该类型存在性和其坐标的可信度,不是必须的,可以用点“.”代替。
7) strand:链的正向与负向,分别用加号+和减号-表示。
8) frame:密码子偏移,可以是0、1或2。
9) attributes:必须要有以下两个值:
gene_id value: 表示转录本在基因组上的基因座的唯一的ID。gene_id与value值用空格分开,如果值为空,则表示没有对应的基因。
transcript_id value: 预测的转录本的唯一ID。transcript_id与value值用空格分开,空表示没有转录本。
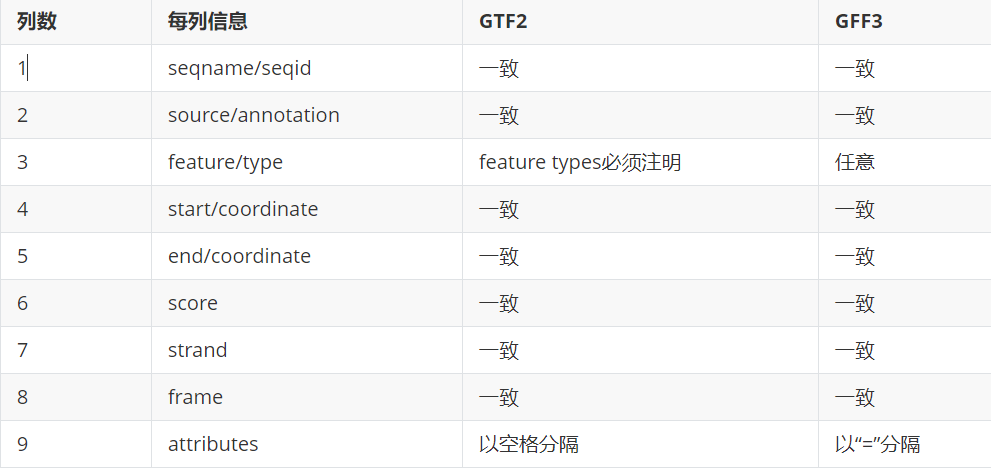
2)GFF3和GTF2之间的异同及相互转换。
---------------------------------------------------
GFF3和GTF2之间的转换可以用Cufflinks里面的工具"gffread":
gffread my.gff3 -T -o my.gtf #gff2gtf
gffread merged.gtf -o- > merged.gff3 #gtf2gff
3) 习题
---------------------------------------
3.1)gff3格式的功能是是什么?目前有几版?
3.2)gff3共有多少列?每一列的含义是什么?
3.3)gff3中的第8列代表的0,1,2分别代表什么含义?
3.4)gff3第9列不同属性之间是用什么符号分割的?
3.5)gtf2和gff3格式上有何异同?
3.6)gtf2和gff3在功能上有什么差异?
3.7)gtf2第9列中不同属性用什么符号分割?
3.8)如何将gtf和gff之间相互转换?
3.9)统计test.gff文件中组装出来的染色体条数
3.10)统计test.gff文件中lnc_RNA个数
3.11)统计基因组文件test.gff中有多少个基因
3.12)求最长基因的长度
3.13)查找一个基因下有3个转录本的基因个数
3.14)求相位为2的cds个数
3.15)找出基因含有最多的外显子的个数
3.16) 将test.gff转化为test.gtf
3.17)统计test.gtf中transcript的个数
3.18)根据test.gtf统计位于正链上的exon的个数
3.19)将test.gtf中所有的gene ID都统计出来
3.20) 找出test.gtf中位于正链上的最长的基因
4) 参考资源
---------------------------------------
https://en.wikipedia.org/wiki/General_feature_format
http://boyun.sh.cn/bio/?p=1602
gff/gtf格式的更多相关文章
- 探索gff/gtf格式
参考: GFF格式说明 Generic Feature Format Version 3 (GFF3) 先下载一个 gtf 文件浏览一下 1 havana gene 11869 14409 . + . ...
- (转) gffcompare和gffread | gtf | gff3 格式文件的分析 | gtf处理 | gtfparse
工具推荐:https://github.com/openvax/gtfparse 真不敢相信,Linux自带的命令会这么强大,从gtf中提取出需要的transcript,看起来复杂,其实一个grep就 ...
- GTF/GFF文件的差异及其相互转换
我们在做生物分析的时候,经常会碰到GFF格式的文件以及GTF格式的注释文件.他们有着相似的名字,甚至连内容都极为相似~那么,他们究竟差在哪里呢? GFF全称为general feature forma ...
- 关于基因组注释文件GTF的解释
GTF文件的全称是gene transfer format,主要是对染色体上的基因进行标注.怎么理解呢,其实所谓的基因名,基因座等,都只是后来人们给一段DNA序列起的名字而已,还原到细胞中就是细胞核里 ...
- 如何使用SnpEff 对SNP结果进行分析
SnpEff is a variant annotation and effect prediction tool. It annotates and predicts the effects of ...
- 【转录组入门】6:reads计数
作业要求: 实现这个功能的软件也很多,还是烦请大家先自己搜索几个教程,入门请统一用htseq-count,对每个样本都会输出一个表达量文件. 需要用脚本合并所有的样本为表达矩阵.参考:生信编程直播第四 ...
- tophat的用法
概述:tophat是以bowtie2为核心的一款比对软件. tophat工作分两步: 1.将reads用bowtie比对到参考基因组上. 2.将unmapped-reads打断成更小的fragment ...
- Augustus指南(Trainning部分)
Augustus指南 官方 Tutorial Index Augustus是一个真核生物基因预测软件,目前有网页服务端和本地版,它基于Hidden-Markov Model(隐马尔科夫链模型HMM)( ...
- bedtools 每天都会用到的工具
详细的使用说明:http://bedtools.readthedocs.org/en/latest/ Collectively, the bedtools utilities are a swiss- ...
随机推荐
- JavaScript之图片操作2
在前一次,我们实现最简单的图片切换效果,这一次,依旧还是图片切换,具体效果如下: 通过点击下面的小图,上面的大图和标题随之切换.因此,我们需要三个容器分别放标题,大图和小图. <!--大图描述- ...
- onload属性使用方法
onload事件属性是页面的图片文字等全部加载完毕后执行的事件 window.onload=fun1;function fun1(){ document.getElementsByTagName ...
- [转]C# 使用代理访问网络
本文部分内容来自:https://zhidao.baidu.com/question/563196409.html 也可以参考:http://www.cnblogs.com/stuart/p/5442 ...
- [UE4]蓝图转换成C++代码
版本:4.12 1.进行如下设置 2.将项目打包出来(任意一平台都行,本文以Windows为例) 3.打包完成后才会在原项目工程中生成蓝图转换成c++的代码 4.如图路径(转换后的代码路径较深所以一步 ...
- http://www.cnblogs.com/TankXiao/archive/2012/02/06/2337728.html
http://www.cnblogs.com/TankXiao/archive/2012/02/06/2337728.html
- C#中char空值的几种表示方式
C#中char空值的几种表示方式 在C#中char类型的表示方式通常是用单引号作为分隔符,而字符串是用双引号作为分隔符. 例如: 程序代码 程序代码 char a = 'a'; char b = 'b ...
- 【Unix网络编程】chapter3 套接字编程简介
chapter3套接字编程简介3.1 概述 地址转换函数在地址的文本表达和他们存放在套接字地址结构中的二进制值之间进行转换.多数现存的IPv4代码使用inet_addr和inet_ntoa这两个函数, ...
- android官方文档翻译(不断更新中。。。)
最近在自学android,抽空把官方文档的guide跟training差不多看了一遍,又对比了一些书籍,感觉还是官方文档讲得比较好,所以自己计划把官方文档翻译一下,方便自己的知识巩固以及复习查找,由于 ...
- 跳表(skiplist)Python实现
# coding=utf-8 # 跳表的Python实现 import random # 最高层数设置为4 MAX_LEVEL = 4 def randomLevel(): ""& ...
- 代码生成器 CodeSmith 的使用(六)
在上一篇的版本中,我们生成了数据库所有表中的字段,如果要使数据库中的单个表 生成 PetaPoco 构架下的 ORM 映射,使那怎么办.这是这篇博客的主要内容. 首先来看完整的 Camel 规则模板: ...