bioawk
https://github.com/lh3/bioawk
1、基本思想
使用:
usage: bioawk [-F fs] [-v var=value] [-c fmt] [-tH] [-f progfile | 'prog'] [file ...]
bioawk基本思想是把组成不同类型的文件(sam、bam、fasta、fastq、vcf)的基本元素封装成变量,直接调用即可。
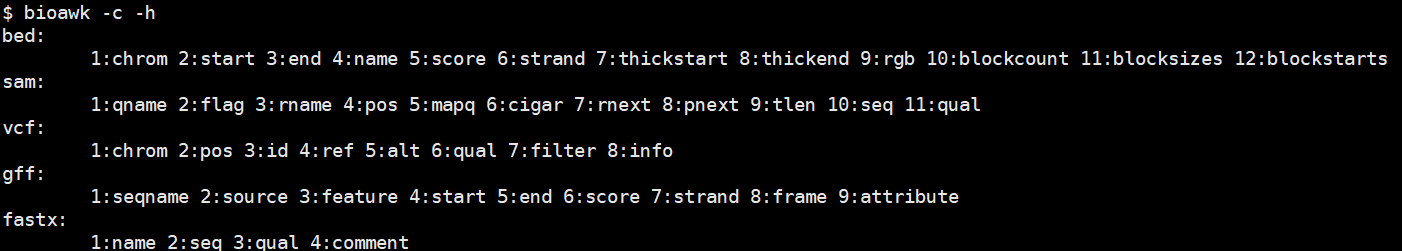
上面出现的名称即可引用其变量。
2、实际例子
打印fasta序列ID、序列、长度、GC含量:
bioawk -c fastx '{print "ID: "$name"\tlength: "length($seq)"\tGC: "gc($seq)"\t"$seq}' demo.fa
引用:https://blog.csdn.net/qq_42491125/article/details/92849378
bioawk的更多相关文章
- 使用bioawk对基因组fasta序列ID(染色体/scaffold名称)排序?
目录 需求 实现 需求 已知某基因组序列,染色体或scaffold ID顺序不定,想要对其按数字排序. 原顺序: 想要的排序结果: 实现 使用bioawk,没有的话conda直接安装. bioawk ...
- Linux command line exercises for NGS data processing
by Umer Zeeshan Ijaz The purpose of this tutorial is to introduce students to the frequently used to ...
随机推荐
- python程序封装成exe流程
在学习python的过程中,在IDE编写完成py项目,运行成功想要封装成exe,方便分享给不同的人即使别人没有安装python也可以使用. 封装的过程中遇到一些问题,记录一下,方便自己和他人查阅. 以 ...
- C语言验证哥德巴赫猜想
#include<stdio.h>int f(int x);int main(void){ int n,i; scanf("%d",&n); for( ...
- antd配置config-overrides.js文件
下载antd 包 npm install antd 下载依赖包(定义组件按需求打包) npm install react-app-rewired customize-cra babel-plugin- ...
- php strlen和mb_strlen
结果: 结论:如果没有中文,尽量使用strlen
- 1+x证书web前端开发jquery专项练习测试题
javascript程序设计-题库 1.下面哪一种不属于Jquery的选择器? A. 基本选择器 B. 层次选择器 C. 表单选择器 D. 节点选择器 答案: D 2.如果需要匹配包含文本的元素,用下 ...
- jmeter进行接口测试--csv参数化,数据驱动-转
首先我们要有一个接口测试用例存放的地方,我们这里用EXCEL模板管理,里面包含用例编号.入参.优先级.请求方式.url等等. 1:新建一个txt文件,命名为sjqd,后缀名改为csv,右键excel格 ...
- [04]ASP.NET Core Web 项目文件
ASP.NET Core Web 项目文件 本文作者:梁桐铭- 微软最有价值专家(Microsoft MVP) 文章会随着版本进行更新,关注我获取最新版本 本文出自<从零开始学 ASP.NET ...
- Caused by: org.springframework.data.mapping.PropertyReferenceException: No property id found for type Users!
Spring Data JPA自定义Repository Caused by: org.springframework.data.mapping.PropertyReferenceException: ...
- WPF图片,DataGrid等实现圆角
<Grid HorizontalAlignment="Center" VerticalAlignment="Center"> <Grid.Ro ...
- C# - WinFrm应用程序MessageBox自动关闭小实验
概述 在程序中MessageBox弹出的对话框,用于向用户展示消息,这是一个模式窗口,可阻止应用程序中的其他操作,直到用户将其关闭.但是有时候在自动化程序中,如果弹出对话框,程序将会中断,等待人工的干 ...