单细胞分析实录(5): Seurat标准流程
前面我们已经学习了单细胞转录组分析的:使用Cell Ranger得到表达矩阵和doublet检测,今天我们开始Seurat标准流程的学习。这一部分的内容,网上有很多帖子,基本上都是把Seurat官网PBMC的例子重复一遍,这回我换一个数据集,细胞类型更多,同时也会加入一些实际分析中很有用的技巧。
1. 导入数据,创建Seurat对象
library(Seurat)
library(tidyverse)
testdf=read.table("test_20210105.txt",header = T,row.names = 1)
test.seu=CreateSeuratObject(counts = testdf)
看一下长什么样子
> test.seu
An object of class Seurat
33538 features across 6746 samples within 1 assay
Active assay: RNA (33538 features)
测试数据有33538个基因,6746个细胞。除此之外,还要关注一下另外两层信息:test.seu@meta.data这个数据框用来存储元数据,每一个细胞都有多个属性;test.seu[["RNA"]]@counts这个稀疏矩阵用来存储原始UMI表达矩阵。
> head(test.seu@meta.data)
orig.ident nCount_RNA nFeature_RNA
A_AAACCCAAGGGTCACA A 3714 1151
A_AAACCCAAGTATAACG A 1855 816
A_AAACCCAGTCTCTCAC A 1530 823
A_AAACCCAGTGAGTCAG A 11145 1087
A_AAACCCAGTGGCACTC A 2289 834
A_AAACGAAAGCCAGAGT A 3714 990
> test.seu[["RNA"]]@counts[1:4,1:4]
4 x 4 sparse Matrix of class "dgCMatrix"
A_AAACCCAAGGGTCACA A_AAACCCAAGTATAACG A_AAACCCAGTCTCTCAC A_AAACCCAGTGAGTCAG
MIR1302-2HG . . . .
FAM138A . . . .
OR4F5 . . . .
AL627309.1 . . . .
2. 简单过滤
接下来,我们根据每个细胞内部线粒体基因表达占比、检测到的基因数、检测的UMI总数这三个方面来对细胞进行简单的过滤。
先计算细胞内线粒体基因表达占比,类似的核糖体基因(大多为RP开头)也能这样计算,还要注意不要将线粒体基因的MT-写成了MT,不然就把别的基因也算进去了:
test.seu[["percent.mt"]] <- PercentageFeatureSet(test.seu, pattern = "^MT-") #正则表达式,表示以MT-开头;test.seu[["percent.mt"]]这种写法会在meta.data矩阵加上一列
这里我已经根据预先设定好的阈值过滤了,代码如下
test.seu <- subset(test.seu, subset = nCount_RNA > 1000 &
nFeature_RNA < 5000 &
percent.mt < 30 &
nFeature_RNA > 600)
过滤之后的数值分布如下,用到VlnPlot()函数,该绘图函数里面的feature参数可以是meta.data矩阵的某一列,也可以是某一个基因,很多文章都用这种图展示marker gene
VlnPlot(test.seu,features = c("nCount_RNA", "nFeature_RNA", "percent.mt"))

nFeature_RNA/nCount_RNA不能太小(空液滴),不能太大(doublet、测序技术限制), 而且阈值设定要综合多个样本来看,像下面这样
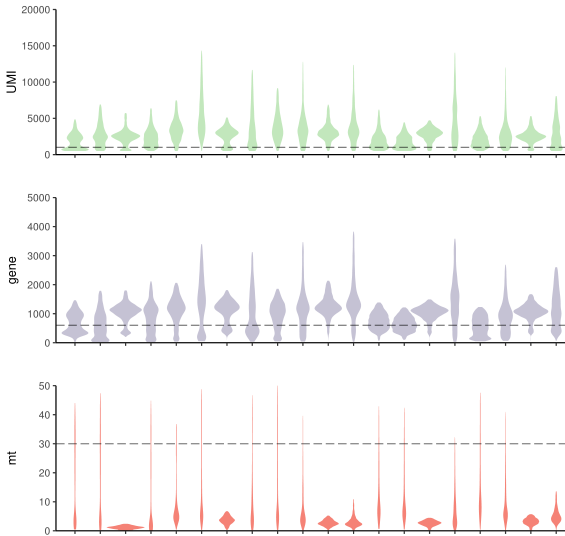
一般在CD45阴性的细胞中percent.mt的阈值大一些,50%也看过几次了
3. 标准化,消除文库大小的影响
如何标准化:LogNormalize: Feature counts for each cell are divided by the total counts for that cell and multiplied by the scale.factor. This is then natural-log transformed using log1p.(先相除,再求对数)
test.seu <- NormalizeData(test.seu, normalization.method = "LogNormalize", scale.factor = 10000)
标准化之后的矩阵存储在test.seu[["RNA"]]@data
> test.seu[["RNA"]]@data[1:4,1:4]
4 x 4 sparse Matrix of class "dgCMatrix"
A_AAACCCAAGGGTCACA A_AAACCCAAGTATAACG A_AAACCCAGTCTCTCAC A_AAACCCAGTGAGTCAG
MIR1302-2HG . . . .
FAM138A . . . .
OR4F5 . . . .
AL627309.1 . . . .
4. 找Variable基因
因为单细胞表达矩阵很稀疏(很多0),选high variable基因的目的可以找到包含信息最多的基因(很多基因的表达差不多都是0),同时极大提升软件运行速度
test.seu <- FindVariableFeatures(test.seu, selection.method = "vst", nfeatures = 2000)
这些基因存储在VariableFeatures(test.seu),有时候可能需要人为指定high variable基因,可以这样:
VariableFeatures(test.seu)="specific genes"
5. 归一化表达矩阵
(基于前面得到的data矩阵)
这一步之后,所有基因的表达值的分布就差不多了,不然表达值不在一个数量级,对后续降维聚类影响挺大。新的矩阵存储在test.seu[["RNA"]]@scale.data里面。
test.seu <- ScaleData(test.seu, features = rownames(test.seu))
默认只对上一步选出来的基因scale,这里调整为所有基因,是为了方便以后画热图,画热图一般会用scale之后的z-score
6. 降维聚类
(基于前面得到的high variable基因的scale矩阵)
test.seu <- RunPCA(test.seu, npcs = 50, verbose = FALSE)
test.seu <- FindNeighbors(test.seu, dims = 1:30)
test.seu <- FindClusters(test.seu, resolution = 0.5)
test.seu <- RunUMAP(test.seu, dims = 1:30)
test.seu <- RunTSNE(test.seu, dims = 1:30)
Run开头的函数降维,Find开头的函数聚类,一般就这几步,相对固定。PCA将原来2000维的数据降到50维,dims参数表示使用多少个主成分(一般20左右就可以了,多几个少几个对结果影响不大),resolution参数表达聚类的分辨率,这个值大于0,一般都是在0-1范围里面调整,越大得到的cluster越多,这个值可以反复调整,并不会改变降维的结果(也就是tsne、umap图的二维坐标)。
这一步之后的数据是这样的
> test.seu
An object of class Seurat
33538 features across 6746 samples within 1 assay
Active assay: RNA (33538 features)
3 dimensional reductions calculated: pca, umap, tsne
# 几种降维方式都会呈现出来
聚类之后多了两列,RNA_snn_res.0.5记录了你用的分辨率,最终的聚类结果保存在seurat_clusters中
> head(test.seu@meta.data)
orig.ident nCount_RNA nFeature_RNA percent.mt RNA_snn_res.0.5
A_AAACCCAAGGGTCACA A 3714 1151 9.585353 8
A_AAACCCAAGTATAACG A 1855 816 12.776280 0
A_AAACCCAGTCTCTCAC A 1530 823 14.248366 12
A_AAACCCAGTGAGTCAG A 11145 1087 2.853297 4
A_AAACCCAGTGGCACTC A 2289 834 15.640017 3
A_AAACGAAAGCCAGAGT A 3714 990 5.654281 0
seurat_clusters
A_AAACCCAAGGGTCACA 8
A_AAACCCAAGTATAACG 0
A_AAACCCAGTCTCTCAC 12
A_AAACCCAGTGAGTCAG 4
A_AAACCCAGTGGCACTC 3
A_AAACGAAAGCCAGAGT 0
7. tsne/umap展示结果
library(cowplot)
test.seu$patient=str_replace(test.seu$orig.ident,"_.*$","")
p1 <- DimPlot(test.seu, reduction = "tsne", group.by = "patient", pt.size=0.5)
p2 <- DimPlot(test.seu, reduction = "tsne", group.by = "ident", pt.size=0.5, label = TRUE,repel = TRUE) #后面两个参数用来添加文本标签
p3 <- DimPlot(test.seu, reduction = "umap", group.by = "patient", pt.size=0.5)
p4 <- DimPlot(test.seu, reduction = "umap", group.by = "ident", pt.size=0.5, label = TRUE,repel = TRUE)
fig_tsne <- plot_grid(p1, p2, labels = c('patient','ident'),align = "v",ncol = 2)
ggsave(filename = "tsne.pdf", plot = fig_tsne, device = 'pdf', width = 30, height = 15, units = 'cm')
fig_umap <- plot_grid(p3, p4, labels = c('patient','ident'),align = "v",ncol = 2)
ggsave(filename = "umap.pdf", plot = fig_umap, device = 'pdf', width = 30, height = 15, units = 'cm')
ident表示每个细胞的标签,聚类之后就是聚类的结果,在一些特定场景可以更换。
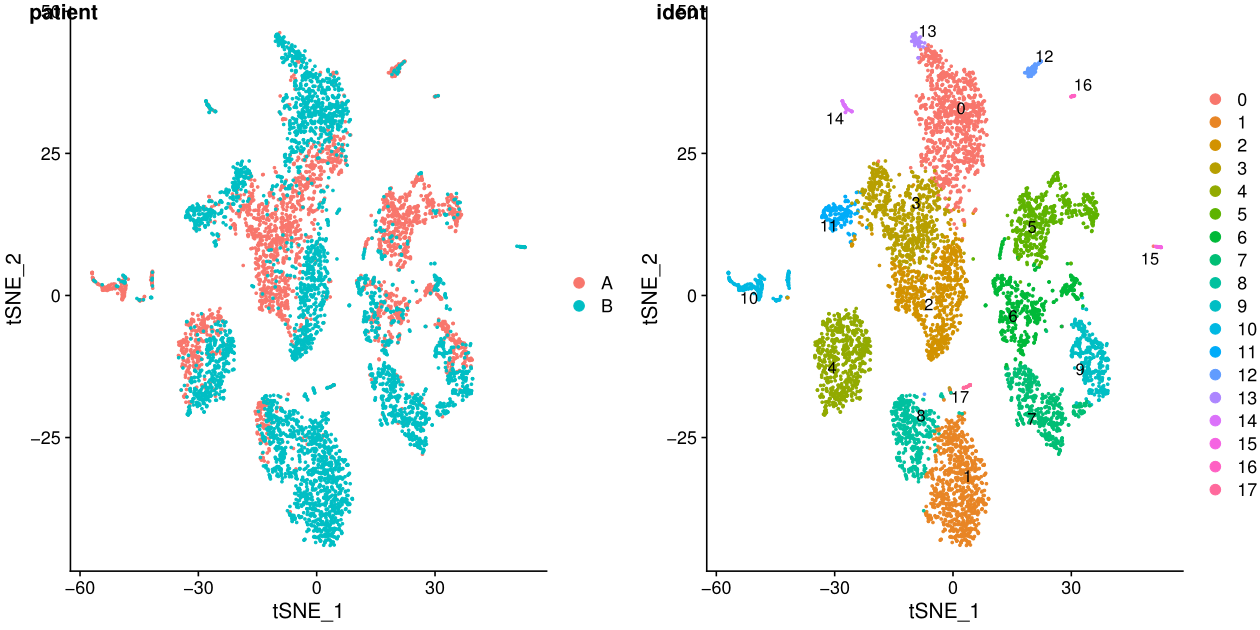
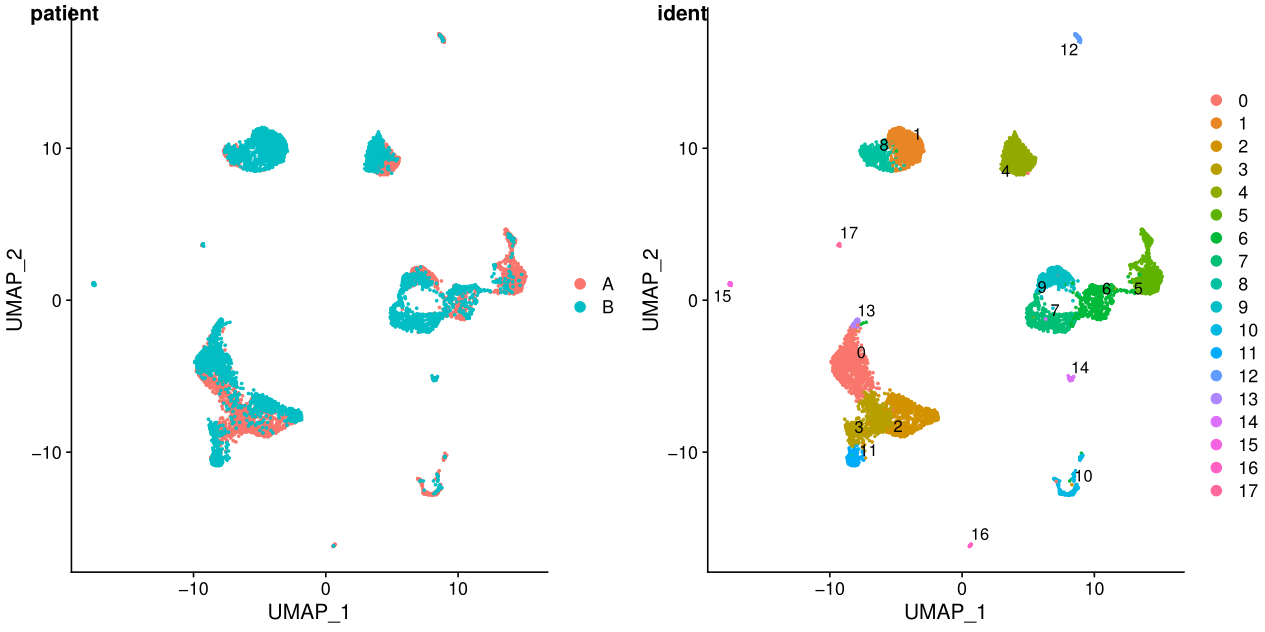
在umap图中,cluster之间的距离更明显
从上面的图可以看出不同样本其实是有批次效应的,下一讲我会介绍两种去批次效应的方法。
因水平有限,有错误的地方,欢迎批评指正!
单细胞分析实录(5): Seurat标准流程的更多相关文章
- 【代码更新】单细胞分析实录(20): 将多个样本的CNV定位到染色体臂,并画热图
之前写过三篇和CNV相关的帖子,如果你做肿瘤单细胞转录组,大概率看过: 单细胞分析实录(11): inferCNV的基本用法 单细胞分析实录(12): 如何推断肿瘤细胞 单细胞分析实录(13): in ...
- 【代码更新】单细胞分析实录(21): 非负矩阵分解(NMF)的R代码实现,只需两步,啥图都有
1. 起因 之前的代码(单细胞分析实录(17): 非负矩阵分解(NMF)代码演示)没有涉及到python语法,只有4个python命令行,就跟Linux下面的ls grep一样的.然鹅,有几个小伙伴不 ...
- 单细胞分析实录(4): doublet检测
最近Cell Systems杂志发表了一篇针对现有几种检测单细胞测序doublet的工具的评估文章,系统比较了常见的例如Scrublet.DoubletFinder等工具在检测准确性.计算效率等方面的 ...
- 单细胞分析实录(1): 认识Cell Hashing
这是一个新系列 差不多是一年以前,我定导后没多久,接手了读研后的第一个课题.合作方是医院,和我对接的是一名博一的医学生,最开始两边的老师很排斥常规的单细胞文章思路,即各大类细胞分群.注释.描述,所以起 ...
- 单细胞分析实录(3): Cell Hashing数据拆分
在之前的文章里,我主要讲了如下两个内容:(1) 认识Cell Hashing:(2): 使用Cell Ranger得到表达矩阵.相信大家已经知道了cell hashing与普通10X转录组的差异,以及 ...
- 单细胞分析实录(8): 展示marker基因的4种图形(一)
今天的内容讲讲单细胞文章中经常出现的展示细胞marker的图:tsne/umap图.热图.堆叠小提琴图.气泡图,每个图我都会用两种方法绘制. 使用的数据来自文献:Single-cell transcr ...
- 单细胞分析实录(17): 非负矩阵分解(NMF)代码演示
本次演示使用的数据来自2017年发表于Cell的头颈鳞癌单细胞文章:Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumo ...
- 单细胞分析实录(2): 使用Cell Ranger得到表达矩阵
Cell Ranger是一个"傻瓜"软件,你只需提供原始的fastq文件,它就会返回feature-barcode表达矩阵.为啥不说是gene-cell,举个例子,cell has ...
- 单细胞分析实录(9): 展示marker基因的4种图形(二)
在上一篇中,我已经讲解了展示marker基因的前两种图形,分别是tsne/umap图.热图,感兴趣的读者可以回顾一下.这一节我们继续学习堆叠小提琴图和气泡图. 3. 堆叠小提琴图展示marker基因 ...
随机推荐
- PyQt(Python+Qt)学习随笔:QTabWidget部件选项卡可用状态访问方法isTabEnabled、setTabEnabled
老猿Python博文目录 专栏:使用PyQt开发图形界面Python应用 老猿Python博客地址 QTabWidget的每个选项卡及页面可设置是否可用,如果选项卡不可用时,则不能通过操作手工切换到该 ...
- Metasploit魔鬼训练营第一章作业
1, Samba服务 Samba是在Linux和UNIX系统上实现SMB协议的一个免费软件,由服务器及客户端程序构成.SMB(Server Messages Block,信息服务块)是一种在局域网上共 ...
- tcp socket学习
更新一波学的socket编程,socket还是比较重要的,探测端口,连接服务底层都是socket编程.tcp有server 和 client.client和udp发送差不多. server端是建立了两 ...
- 学习tcp和udp
tcp和udp的差别 tcp和udp是不一样的 tcp是一个可靠的连接,如果发出一个tcp请求,那么就一定要回复一个请求 而udp是不需要回复的,就像QQ一样,只要发过来就可以了,不管别人在不在线,发 ...
- ADF 第二篇:使用UI创建ADF
用户可以通过UI来创建ADF,在UI中创建ADF时,用户不需要下载单独的IDE,而仅仅通过 Microsoft Edge 或者 Google Chrome浏览器.用户登录Azure Portal,选择 ...
- 公司只提供签名服务,不提供证书文件,如何打包Electron应用
需求 稍微正规点的公司,都要为自己开发的软件做代码签名,如下图所示 代码签名的主要目的是为了确保软件的来源(这个软件是由谁生产的)和软件的内容不被篡改 一个软件公司可能有很多团队,很多开发者,开发不同 ...
- 廖雪峰官网学习js 数据类型和变量
数据类型: number 不分整数 和浮点数 字符串 用' ' " " 表示 布尔值 true false && 与运算符(都ture才ture ...
- NPM相关知识点
1.Windows环境变量的配置 npm config set prefix "D:\Program Files\nodejs\node_global" npm config se ...
- DBeaver连接MySQ报错
遇错情况:第一次使用DBaver连接MySQL遇到以下问题: 报错信息:Public Key Retrieval is not allowed 截图如下: 解决方案步骤: 一.已有连接的情况:F4或者 ...
- Consul的使用
Consul的使用 生产部署中,Consul安装在要注册服务的每个节点上.Consul有两种运行模式:客户端和服务器端,每个Consul数据中心必须至少有一个服务器,负责维护Consul状态,为了 ...