利用ONT测序检测真核生物全基因组甲基化状态
摘要
甲基化在真核生物基因组序列中广泛存在,其中5mC最为普遍,在真核生物基因组中也有发现6mA。捕获基因组中的甲基化状态的常用技术是全基因组甲基化测序(WGBS)和简化甲基化测序(RRBS),而随着第三代测序技术的完善,ONT单分子纳米孔测序可以从单分子的角度来检出甲基化的胞嘧啶和腺嘌呤电流的变化,从而实现由基因组中的一段序列中检出5mC和6mA,然而精确地从单碱基级别检出5mC和6mA扔具有挑战。本文利用第三代ONT测序技术获得的序列及其电信号来检出真核生物全基因组范围的5mC和6mA甲基化状态。
背景
DNA甲基化主要发生在脱氧核糖核苷酸的第五位的胞嘧啶和第六位的腺嘌呤,前者普遍存在于真核生物,后者在原核生物中广泛存在,也有研究报道6mA存在于真核生物。这样的甲基化状态在ONT测序仪捕捉到的电流信号中,不仅单碱基的电流会发生改变,而且其上下文的一段基因组序列也会发生改变。基于此,一些生物信息学软件先后被开发出来针对于这两种甲基化的检出有各自的优缺点。有研究指出在真核基因组中检出5mC和6mA准确度较高的软件分别为nanopolish[1]和tombo[2]。
利用nanopolish检出真核生物基因组中5mC的甲基化位置
材料和方法
利用minION平台对目标生物血液提取的DNA不打断建库并进行全基因组测序,获得12G序列及其电信号文件。安装nanopolish(v0.13.2)。
步骤
- 建立索引
nanopolish index -d fast5_files/ output.fastq
- 比对
minimap2 -a -x map-ont reference.fasta output.fastq | samtools sort -T tmp -o output.sorted.bam
samtools index output.sorted.bam
- Calling methylation
nanopolish call-methylation --progress -q cpg -t NCPU --verbose -r reads.fastq -b output.sorted.bam -g reference_genome.fasta > nanopolish_call_methylation.tsv
- 筛选高置信度的甲基化和未甲基化位点
calculate_methylation_frequency.py[3]
nanopore-methylation-utilities/mtsv2bedGraph.py[4]
scripts/calculate_methylation_frequency.py -c 2 methylation_calls.tsv > methylation_frequency.tsv
# or
python nanopore-methylation-utilities/parseMethylbed.py frequency -i methylation_calls.tsv -o methylation_calls_freq.tsv --verbose -m cpg -u 2 -l -2
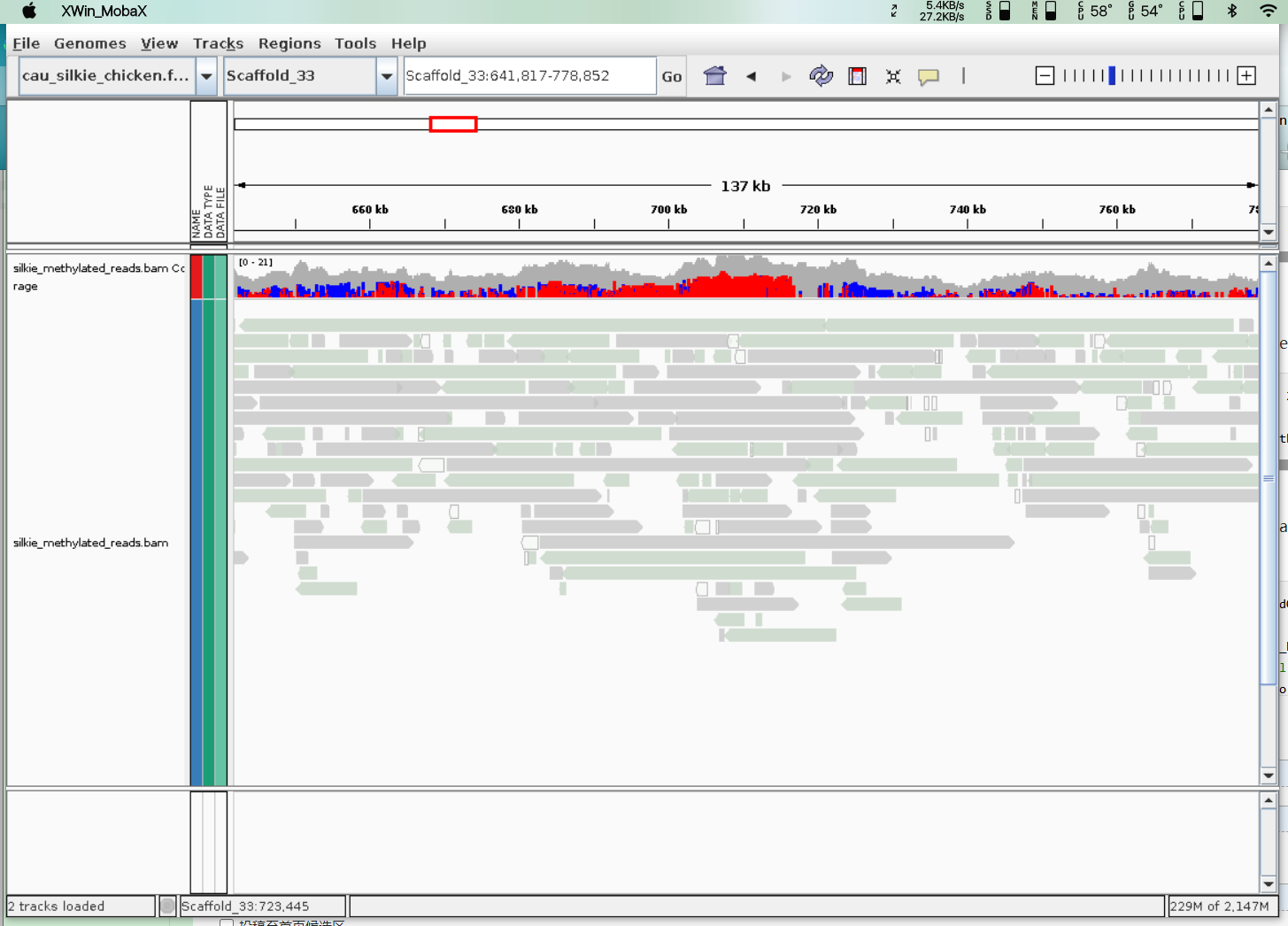
- IGV可视化或UCSC可视化
IGV可视化[5]
python nanopore-methylation-utilities/mtsv2bedGraph.py --verbose -c 2 -i methylation_calls.tsv -q cpg -g reference_genome.fasta | sort -k1,1 -k2,2n | bgzip > methylation_calls.bed.gz
tabix -p methylation_calls.bed.gz
python nanopore-methylation-utilities/convert_bam_for_methylation.py -t 100 --verbose --remove_poor -c methylation_calls.bed.gz -f reference_genome.fasta -b output.sorted.bam | samtools sort -o methylation_calls.bed.remove_no_or_poor_methylation_reads.bam
# if all reads' coverage was needed, remap all reads to reference to find out
samtools index methylation_calls.bed.remove_no_or_poor_methylation_reads.bam
# now bam file can be loaded to igv via their bisulfite mode to see methylation sites and unmethylation sites.
利用ONT测序检测真核生物全基因组甲基化状态的更多相关文章
- 全基因组测序 Whole Genome Sequencing
全基因组测序 Whole Genome Sequencing 全基因组测序(Whole Genome Sequencing,WGS)是利用高通量测序平台对一种生物的基因组中的全部基因进行测序,测定其 ...
- cfDNA(circulating cell free DNA)全基因组测序
参考资料: [cfDNA专题]cell-free DNA在非肿瘤疾病中的临床价值(好) ctDNA, cfDNA和CTCs有什么区别吗? cfDNA你懂多少? 新发现 | 基因是否表达,做个cfDNA ...
- 全基因组测序 从头测序(de novo sequencing) 重测序(re-sequencing)
全基因组测序 全基因组测序分为从头测序(de novo sequencing)和重测序(re-sequencing). 从头测序(de novo)不需要任何参考基因组信息即可对某个物种的基因组进行测序 ...
- PacBio全基因组测序和组装
PacBio公司的业务范围也就5个(官网): Whole Genome Sequencing Targeted Sequencing Complex Populations RNA Sequencin ...
- WGS 全基因组测序数据分析
1. DNA测序技术 https://www.jianshu.com/p/6122cecec54a 2.FASTA和FASTQ文件格式 https://www.jianshu.com/p/50ff30 ...
- GWAS | 全基因组关联分析 | Linkage disequilibrium (LD)连锁不平衡 | 曼哈顿图 Manhattan_plot | QQ_plot | haplotype phasing
现在GWAS已经属于比较古老的技术了,主要是碰到严重的瓶颈了,单纯的snp与表现的关联已经不够,需要具体的生物学解释,这些snp是如何具体导致疾病的发生的. 而且,大多数病找到的都不是个别显著的snp ...
- 如何鉴定全基因组加倍事件(WGD)
目前鉴定全基因组加倍(whole-genome duplication events)有3种 通过染色体共线性(synteny) 方法是比较两个基因组的序列,并将同源序列的位置绘制成点状图,如果能在点 ...
- 【GWAS文献解读】疟原虫青蒿素抗药性的全基因组关联分析
英文名:Genetic architecture of artemisinin-resistant Plasmodium falciparum 中文名:疟原虫青蒿素抗药性的全基因组关联分析 期刊:Na ...
- Genome-wide Complex Trait Analysis(GCTA)-全基因组复杂性状分析
GCTA(全基因组复杂性状分析)工具开发目的是针对复杂性状的全基因组关联分析,评估SNP解释的表型方差所占的比例(该网站地址:http://cnsgenomics.com/software/gcta/ ...
随机推荐
- 变体 variety 计算机学科中的改变类型;输入法的 类型
变体_百度百科 中文为改变原来的体式.或者计算机学科中的改变类型. 变体 variety 输入法的 类型
- 查找目录下的所有文件中是否含有某个字符串 find .|xargs grep -ri "IBM"
linux查看目录下所有文件内容中是否包含某个字符串 2017-07-25 15:13:22 默一鸣 阅读数 21556 文章标签: linux查找文件夹文件内容字符串 更多 分类专栏: Unix ...
- Linux服务之nginx服务篇二(搭建)
一.简易搭建安装步骤 0.检查环境 1.配置yum源 使用yum list nginx 检查yum源中是否有nginx安装包 #官方网络源需要安装epel-* #或使用251的adv源(老师的yum源 ...
- 【转载】java与xml
原文地址:http://www.lai18.com/content/1198237.html java项目中,xml文件一般都是用来存储一些配置信息一般的编程, 多数用来存储配置信息 . 拿JDBC来 ...
- GPIO端口上拉下拉 与 硬件图的上拉下拉
硬件图上的上拉下拉: 没有触发时默认接到IO的是高电平就是上拉: 没有触发时默认接到IO的是低电平就是 下拉: (2)对应GPIO的配置 配置与你的外围电路息息相关: 比如下图: 你只能配置为上拉: ...
- Linux下安装JDK 1.8你必须知道的糟心事
来源:Atstudy网校 1.简介 在Oracle收购Sun后,Java的一系列产品就被整合到Oracle官网中,打开官网乍眼一看也不知道去哪里下载,还的一个一个的摸索尝试,而且网上大多数都是一些Or ...
- 解决Error running 'Tomcat 9': Address localhost:8080 is already in use的问题
在我学习servlet的过程中遇到了tomacat端口8080被占用的情况,所以记录下来,毕竟以后还会碰见这种貌似情况 第一步,打开命令行界面,可快捷键window+R打开输入cmd进入 输入代码:n ...
- 结构感知图像修复:ICCV2019论文解析
结构感知图像修复:ICCV2019论文解析 StructureFlow: Image Inpainting via Structure-aware Appearance Flow 论文链接: http ...
- MySQL面试题汇总
事务是什么? 一系列操作,要么全部完成,要么一个都不做 事务的ACID特性 原子性:一系列操作要么都执行,要么都不执行 一致性:事务执行前后数据完整性不变,如转账前后总金额不变 隔离性:多个事务并发访 ...
- NX二次开发】Block UI 选择特征
属性说明 属性 类型 描述 常规 BlockID String 控件ID Enable Logical 是否可操作 Group ...